

Activation of atypical protein kinase C by sphingosine 1-phosphate revealed by an aPKC-specific activity reporter
- PMID: 30600259
- PMCID: PMC6657501
- DOI: 10.1126/scisignal.aat6662
Activation of atypical protein kinase C by sphingosine 1-phosphate revealed by an aPKC-specific activity reporter
Abstract
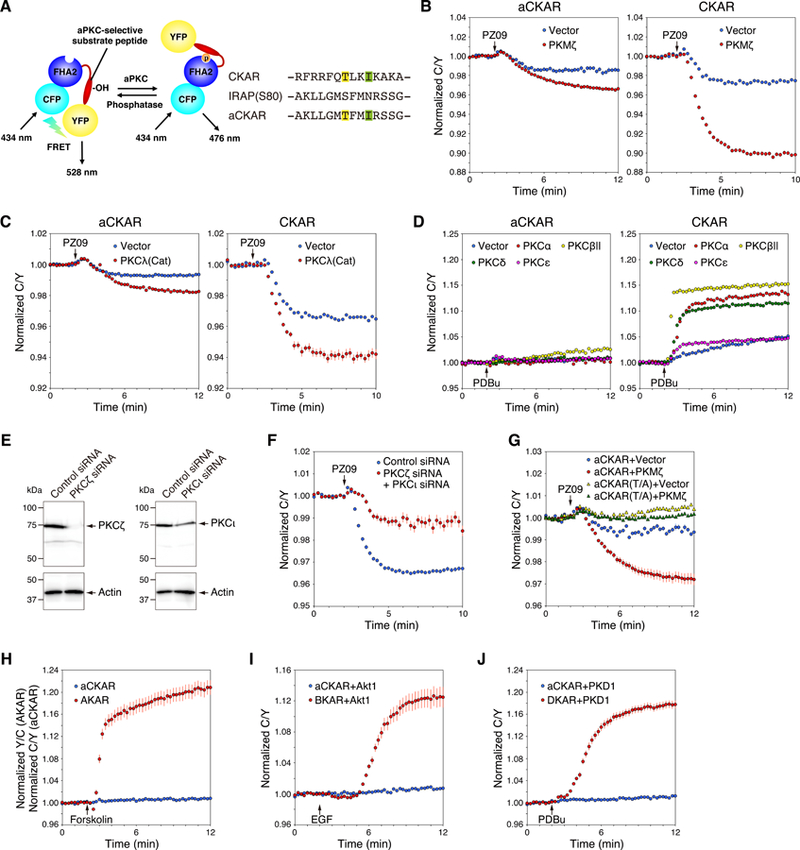
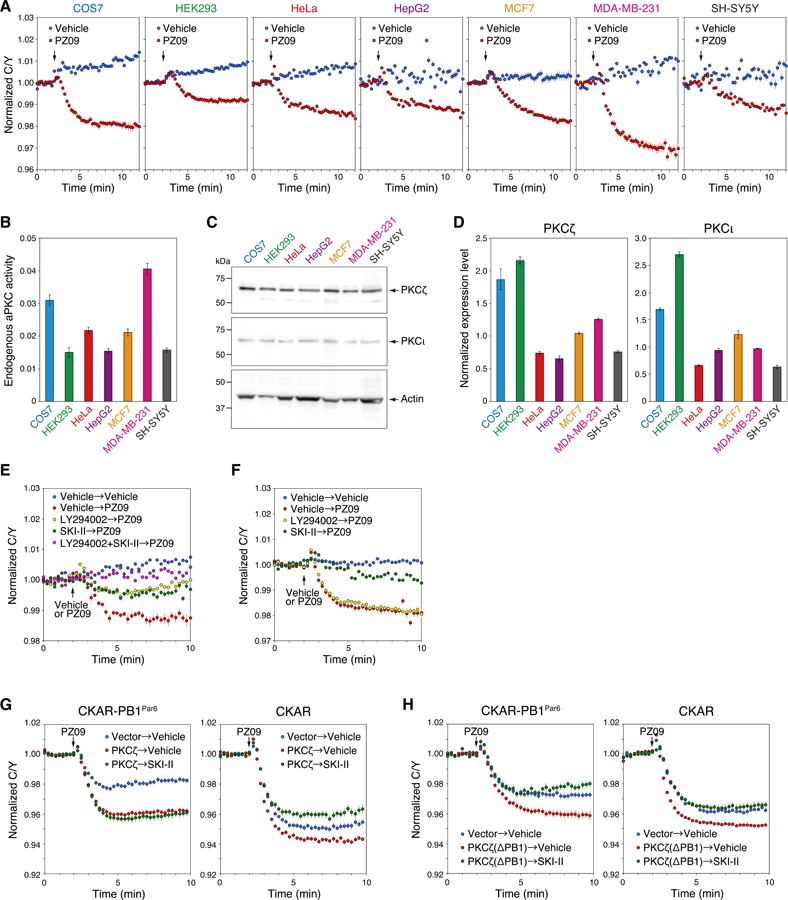
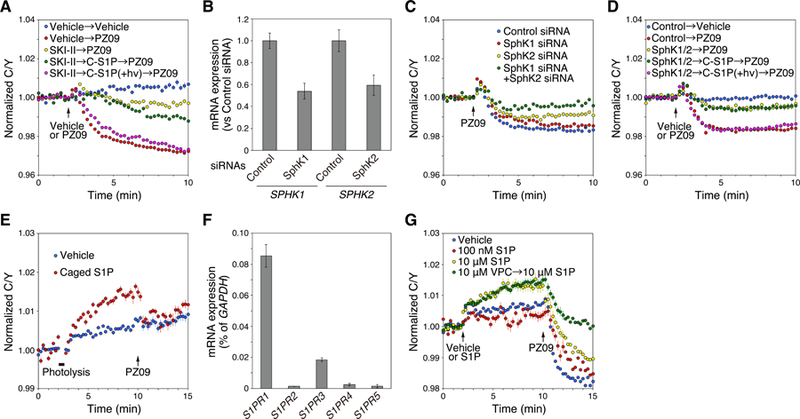
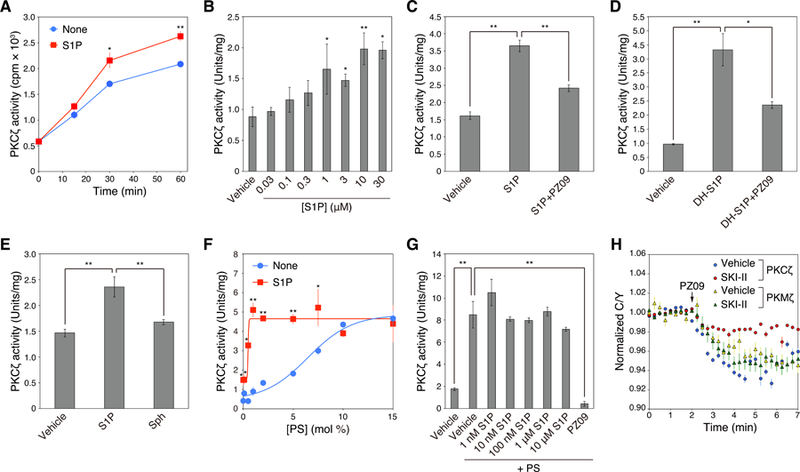
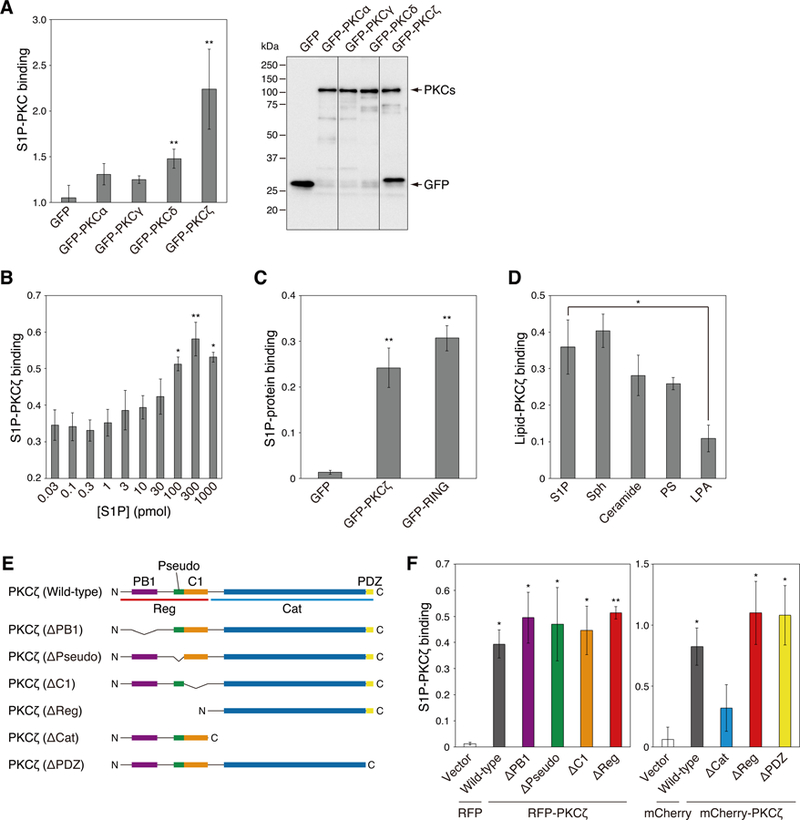
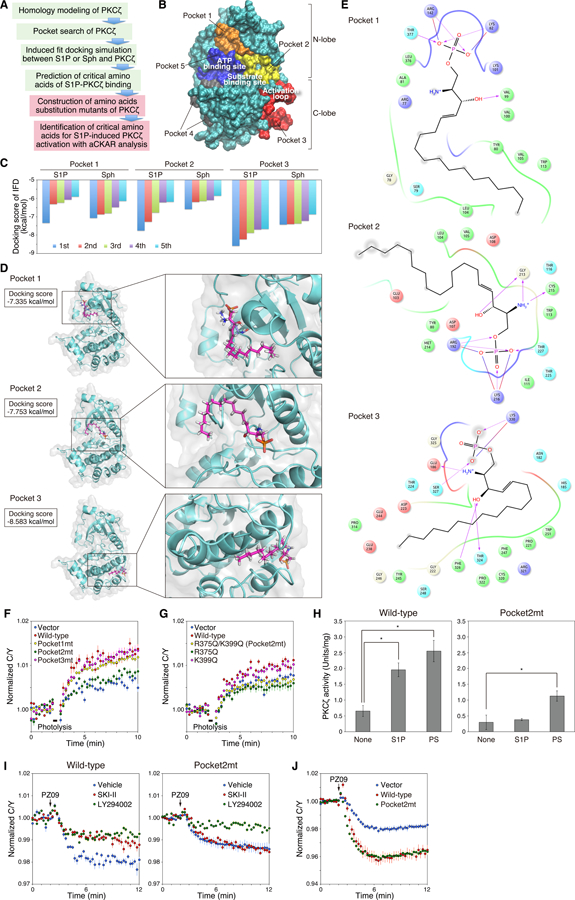
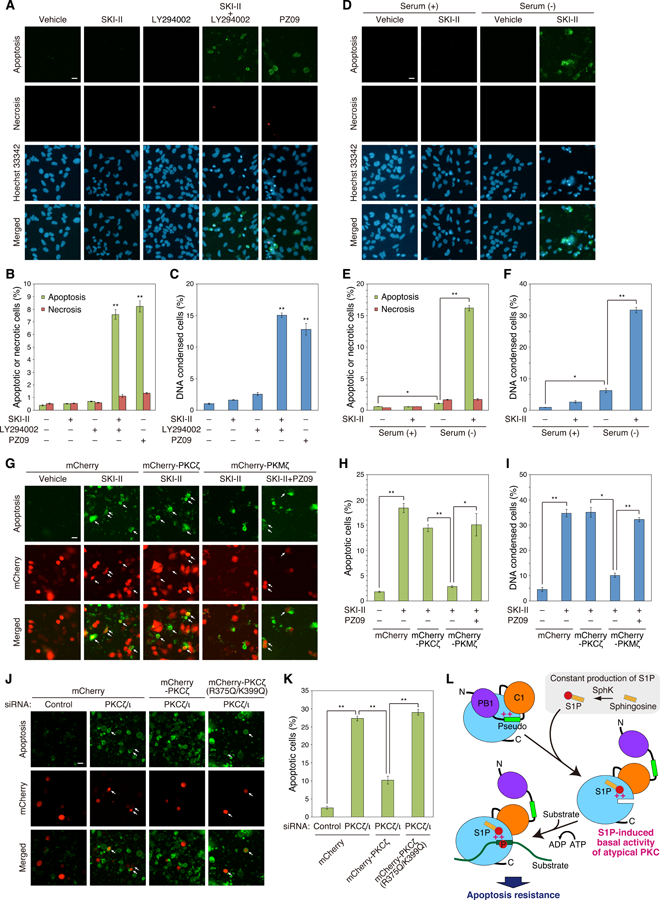
Atypical protein kinase C (aPKC) isozymes are unique in the PKC superfamily in that they are not regulated by the lipid second messenger diacylglycerol, which has led to speculation about whether a different second messenger acutely controls their function. Here, using a genetically encoded reporter that we designed, aPKC-specific C kinase activity reporter (aCKAR), we found that the lipid mediator sphingosine 1-phosphate (S1P) promoted the cellular activity of aPKC. Intracellular S1P directly bound to the purified kinase domain of aPKC and relieved autoinhibitory constraints, thereby activating the kinase. In silico studies identified potential binding sites on the kinase domain, one of which was validated biochemically. In HeLa cells, S1P-dependent activation of aPKC suppressed apoptosis. Together, our findings identify a previously undescribed molecular mechanism of aPKC regulation, a molecular target for S1P in cell survival regulation, and a tool to further explore the biochemical and biological functions of aPKC.
Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
References
-
- Suzuki A, Ohno S, The PAR-aPKC system: lessons in polarity. Journal of cell science 119, 979–987 (2006). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
