

Integration of DNA methylation patterns and genetic variation in human pediatric tissues help inform EWAS design and interpretation
- PMID: 30602389
- PMCID: PMC6314079
- DOI: 10.1186/s13072-018-0245-6
Integration of DNA methylation patterns and genetic variation in human pediatric tissues help inform EWAS design and interpretation
Abstract
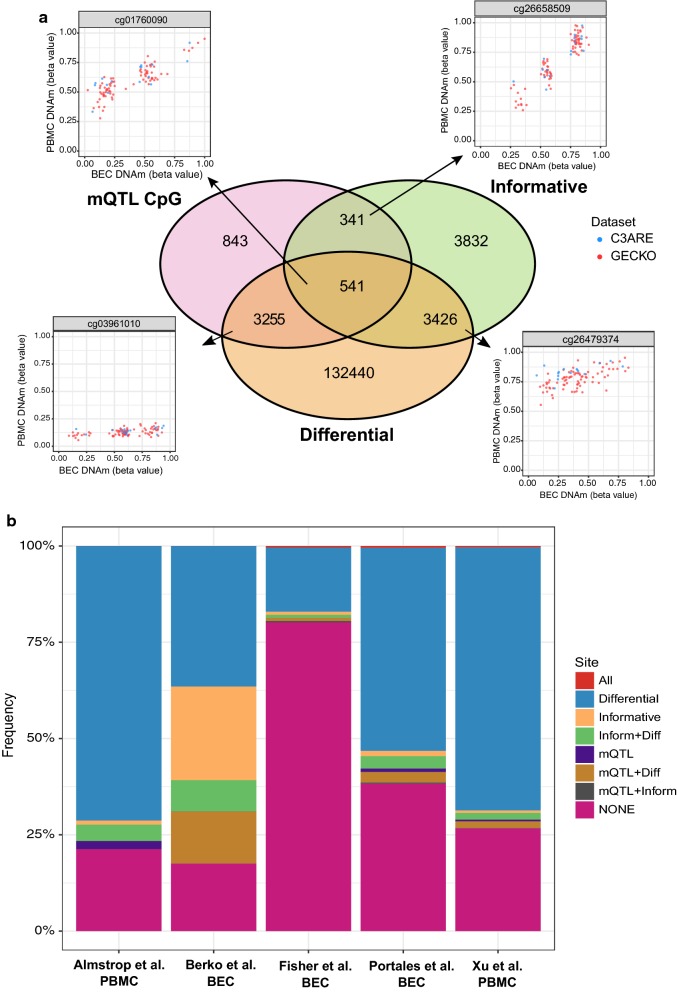
Background: The widespread use of accessible peripheral tissues for epigenetic analyses has prompted increasing interest in the study of tissue-specific DNA methylation (DNAm) variation in human populations. To date, characterizations of inter-individual DNAm variability and DNAm concordance across tissues have been largely performed in adult tissues and therefore are limited in their relevance to DNAm profiles from pediatric samples. Given that DNAm patterns in early life undergo rapid changes and have been linked to a wide range of health outcomes and environmental exposures, direct investigations of tissue-specific DNAm variation in pediatric samples may help inform the design and interpretation of DNAm analyses from early life cohorts. In this study, we present a systematic comparison of genome-wide DNAm patterns between matched pediatric buccal epithelial cells (BECs) and peripheral blood mononuclear cells (PBMCs), two of the most widely used peripheral tissues in human epigenetic studies. Specifically, we assessed DNAm variability, cross-tissue DNAm concordance and genetic determinants of DNAm across two independent early life cohorts encompassing different ages.
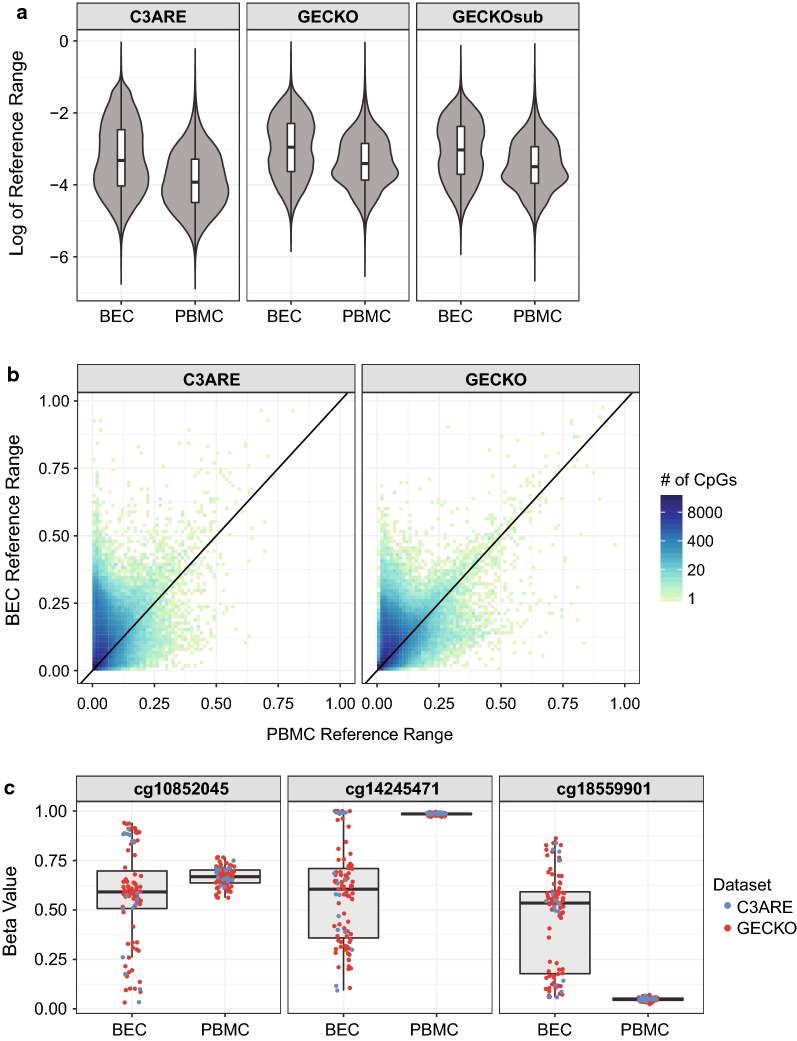
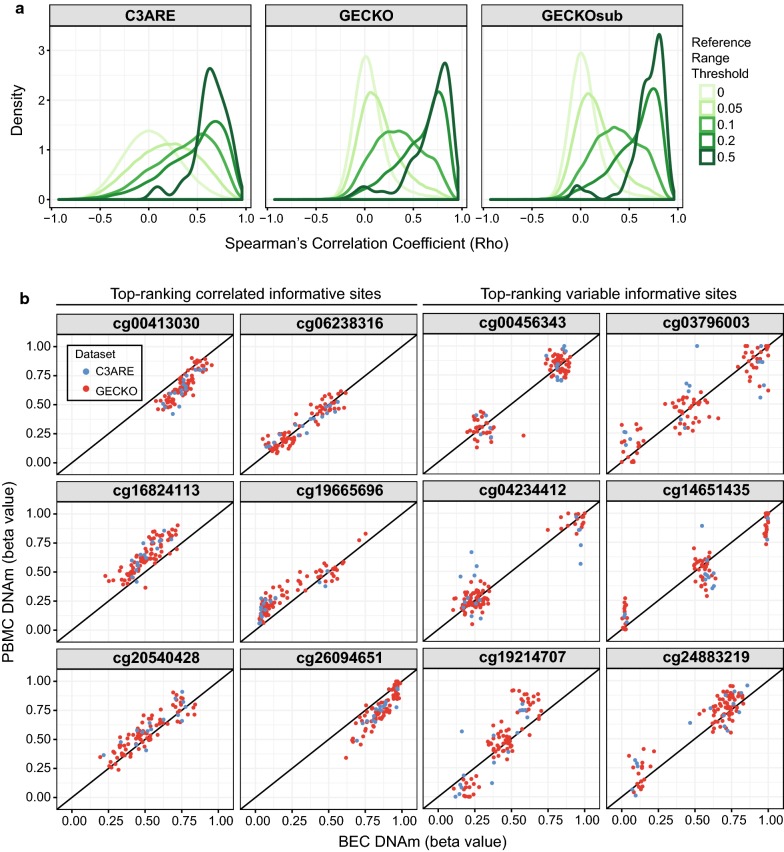
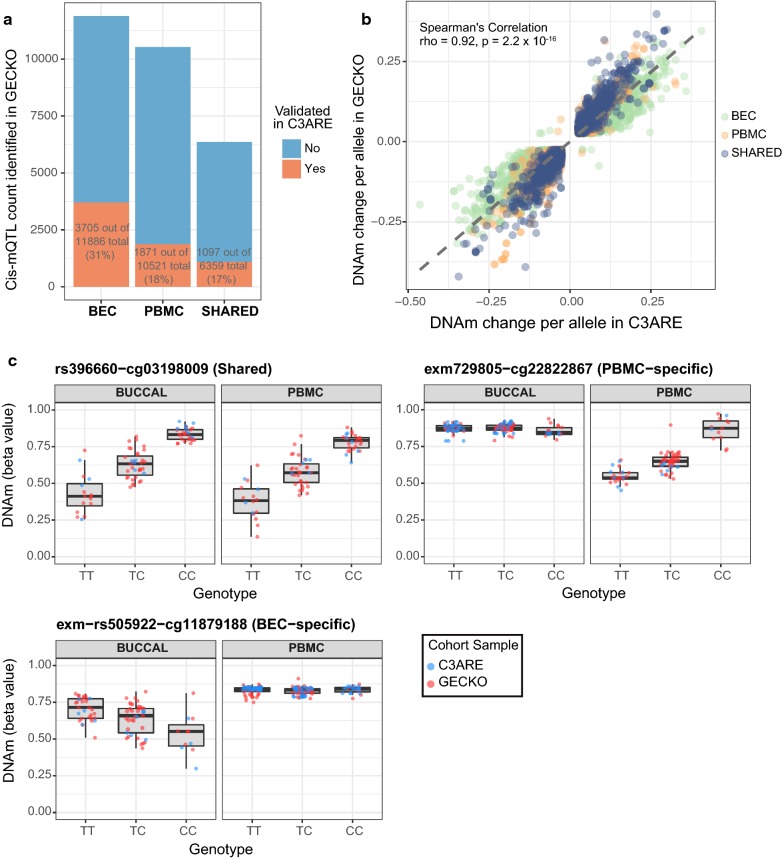
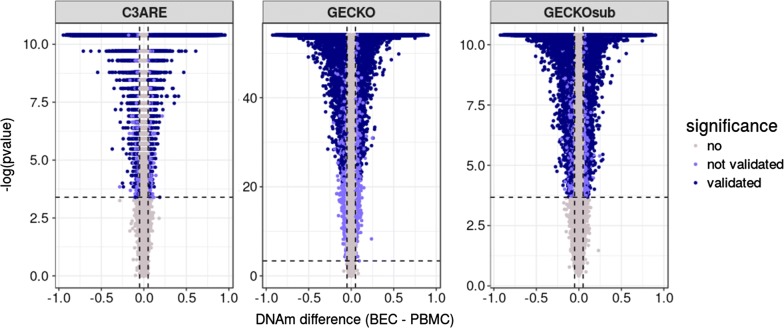
Results: BECs had greater inter-individual DNAm variability compared to PBMCs and highly the variable CpGs are more likely to be positively correlated between the matched tissues compared to less variable CpGs. These sites were enriched for CpGs under genetic influence, suggesting that a substantial proportion of DNAm covariation between tissues can be attributed to genetic variation. Finally, we demonstrated the relevance of our findings to human epigenetic studies by categorizing CpGs from published DNAm association studies of pediatric BECs and peripheral blood.
Conclusions: Taken together, our results highlight a number of important considerations and practical implications in the design and interpretation of EWAS analyses performed in pediatric peripheral tissues.
Keywords: Buccal epithelial cells; DNA methylation; Genetic variation; Illumina 450K array; Pediatric; Peripheral blood leukocytes; Surrogate tissues.
Figures
References
-
- Chadwick LH, Sawa A, Yang IV, Baccarelli A, Breakefield XO, Deng H-WW, et al. New insights and updated guidelines for epigenome-wide association studies. Neuroepigenetics. 2015;1C:14–19. doi: 10.1016/j.nepig.2014.10.004. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
