

HIV peptidome-wide association study reveals patient-specific epitope repertoires associated with HIV control
- PMID: 30602460
- PMCID: PMC6338868
- DOI: 10.1073/pnas.1812548116
HIV peptidome-wide association study reveals patient-specific epitope repertoires associated with HIV control
Abstract
Genetic variation in the peptide-binding groove of the highly polymorphic HLA class I molecules has repeatedly been associated with HIV-1 control and progression to AIDS, accounting for up to 12% of the variation in HIV-1 set point viral load (spVL). This suggests a key role in disease control for HLA presentation of HIV-1 epitopes to cytotoxic T cells. However, a comprehensive understanding of the relevant HLA-bound HIV epitopes is still elusive. Here we describe a peptidome-wide association study (PepWAS) approach that integrates HLA genotypes and spVL data from 6,311 HIV-infected patients to interrogate the entire HIV-1 proteome (3,252 unique peptides) for disease-relevant peptides. This PepWAS approach predicts a core set of epitopes associated with spVL, including known epitopes but also several previously uncharacterized disease-relevant peptides. More important, each patient presents only a small subset of these predicted core epitopes through their individual HLA-A and HLA-B variants. Eventually, the individual differences in these patient-specific epitope repertoires account for the variation in spVL that was previously associated with HLA genetic variation. PepWAS thus enables a comprehensive functional interpretation of the robust but little-understood association between HLA and HIV-1 control, prioritizing a short list of disease-associated epitopes for the development of targeted therapy.
Keywords: HIV-1; HLA; MHC; antigen presentation; epitope prediction.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
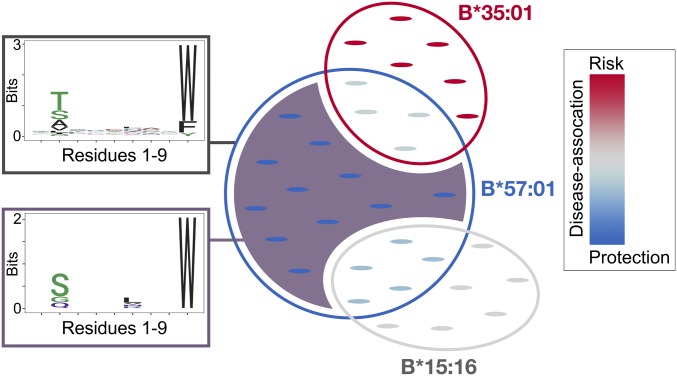
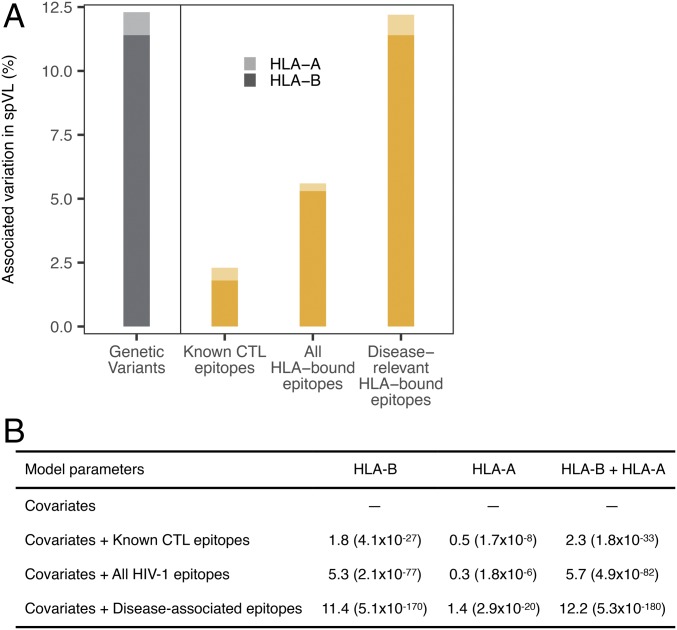
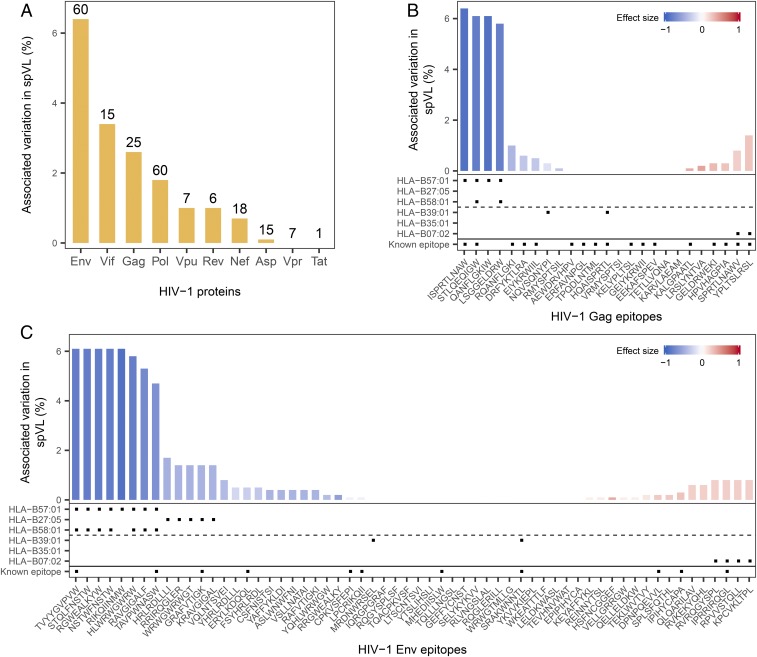
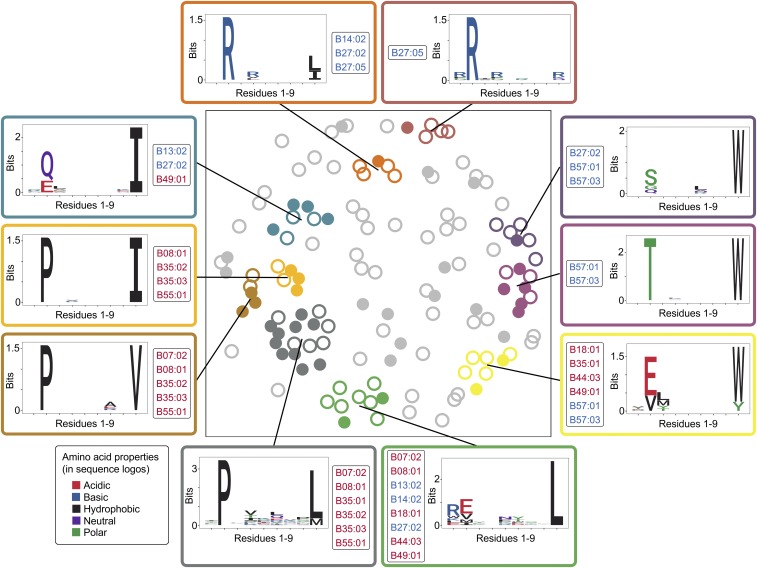
References
-
- Mellors JW, et al. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996;272:1167–1170, and erratum (1997) 275:14. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
