

Single cell RNA analysis identifies cellular heterogeneity and adaptive responses of the lung at birth
- PMID: 30604742
- PMCID: PMC6318311
- DOI: 10.1038/s41467-018-07770-1
Single cell RNA analysis identifies cellular heterogeneity and adaptive responses of the lung at birth
Abstract
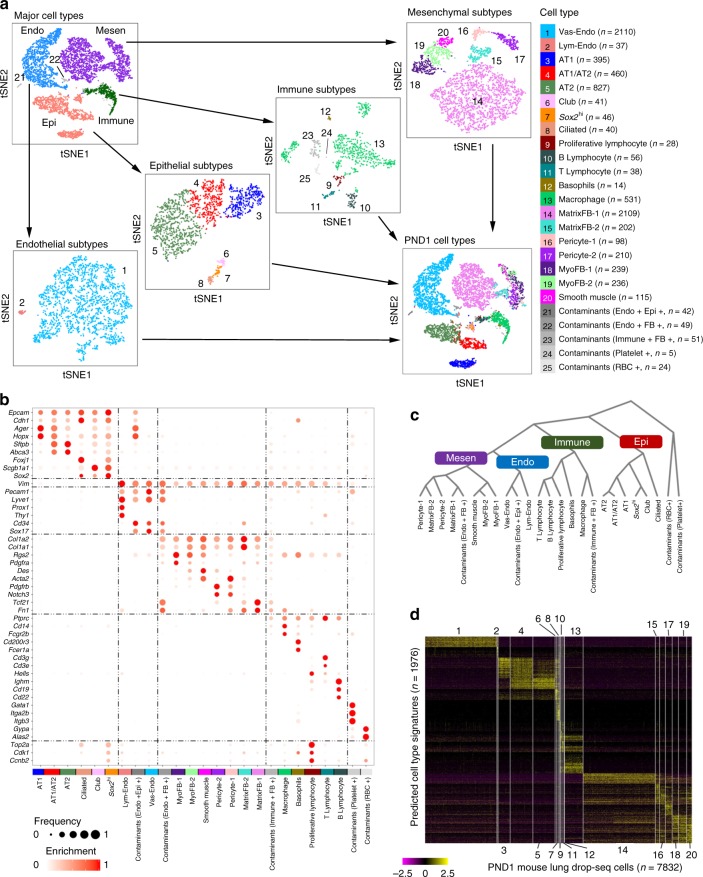
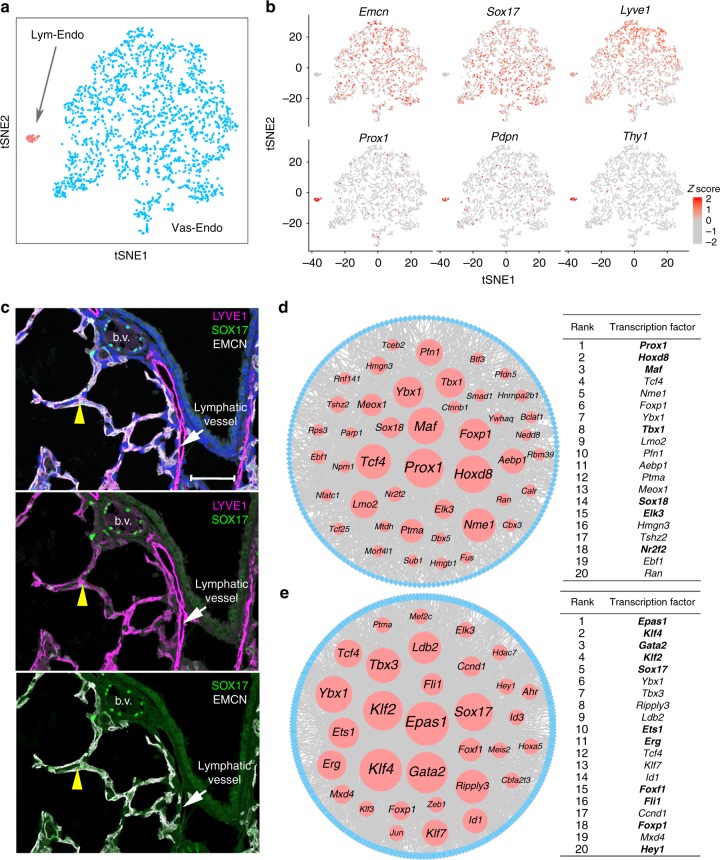
The respiratory system undergoes a diversity of structural, biochemical, and functional changes necessary for adaptation to air breathing at birth. To identify the heterogeneity of pulmonary cell types and dynamic changes in gene expression mediating adaptation to respiration, here we perform single cell RNA analyses of mouse lung on postnatal day 1. Using an iterative cell type identification strategy we unbiasedly identify the heterogeneity of murine pulmonary cell types. We identify distinct populations of epithelial, endothelial, mesenchymal, and immune cells, each containing distinct subpopulations. Furthermore we compare temporal changes in RNA expression patterns before and after birth to identify signaling pathways selectively activated in specific pulmonary cell types, including activation of cell stress and the unfolded protein response during perinatal adaptation of the lung. The present data provide a single cell view of the adaptation to air breathing after birth.
Conflict of interest statement
The authors declare no competing interests.
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
