

Exploring the underlying biology of intrinsic cardiorespiratory fitness through integrative analysis of genomic variants and muscle gene expression profiling
- PMID: 30605401
- PMCID: PMC6589809
- DOI: 10.1152/japplphysiol.00035.2018
Exploring the underlying biology of intrinsic cardiorespiratory fitness through integrative analysis of genomic variants and muscle gene expression profiling
Abstract
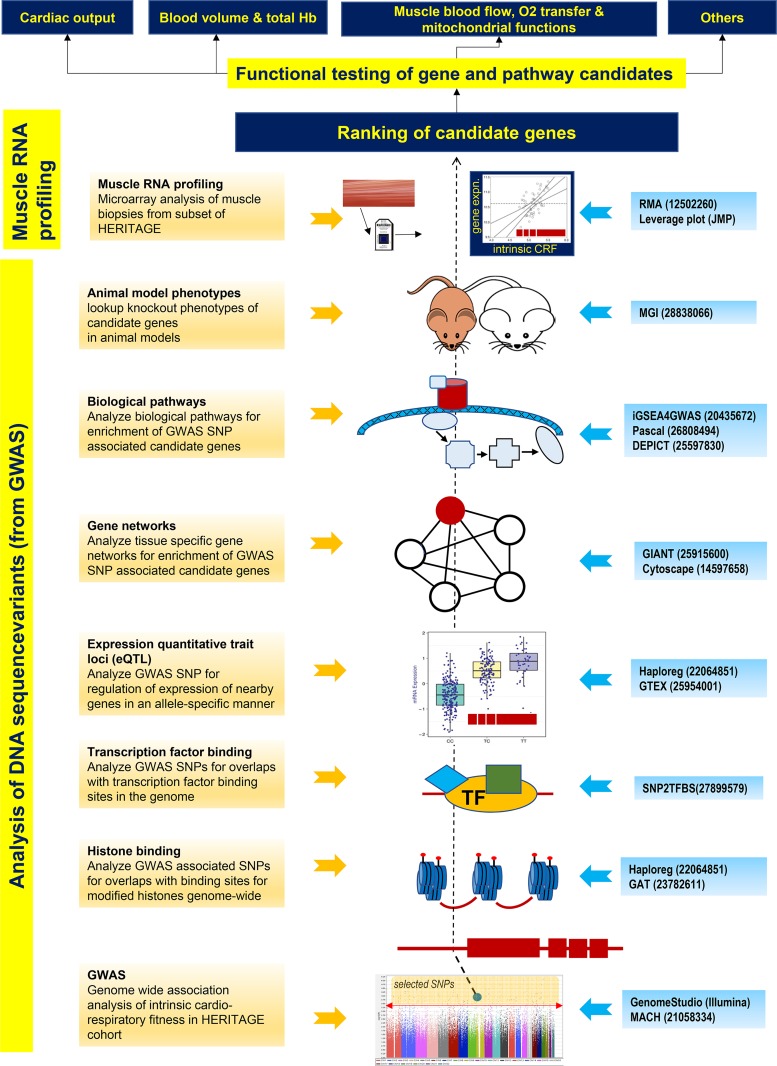
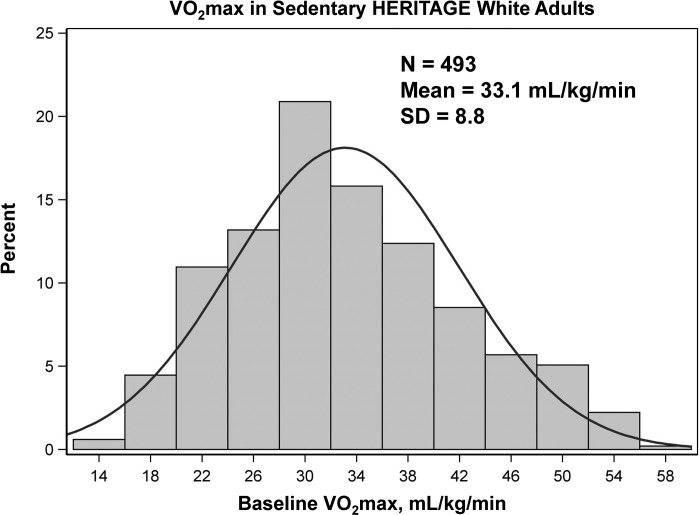
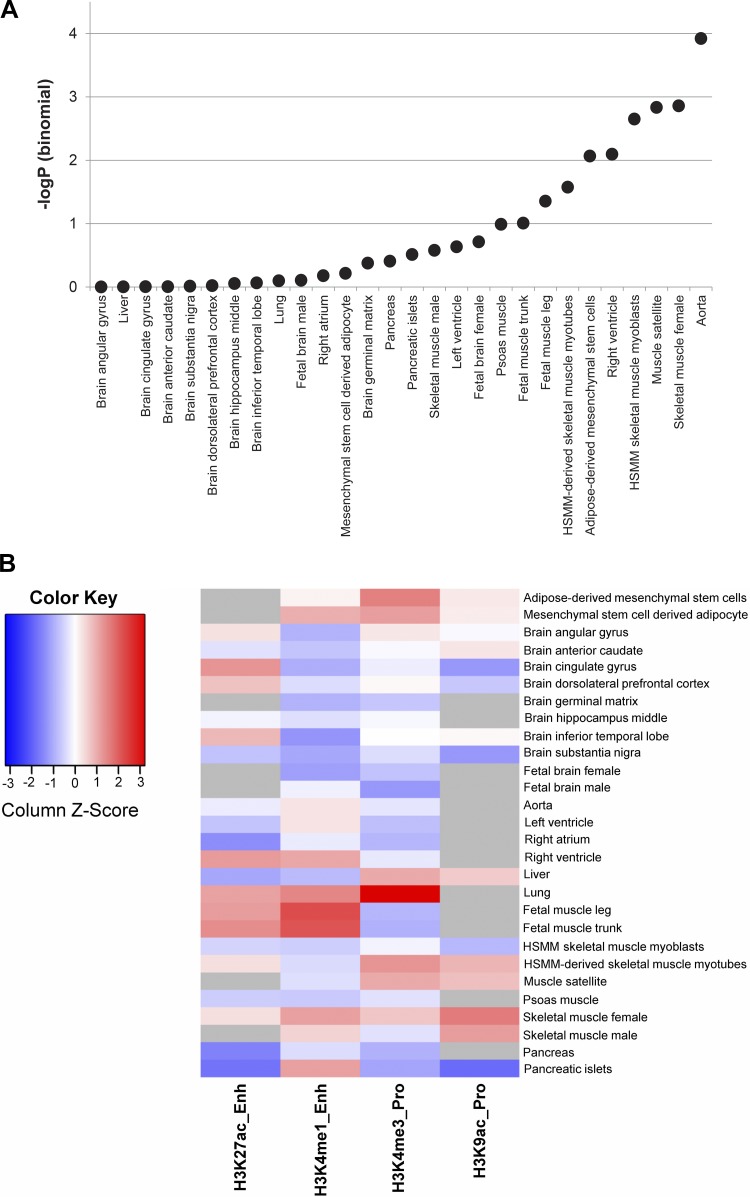
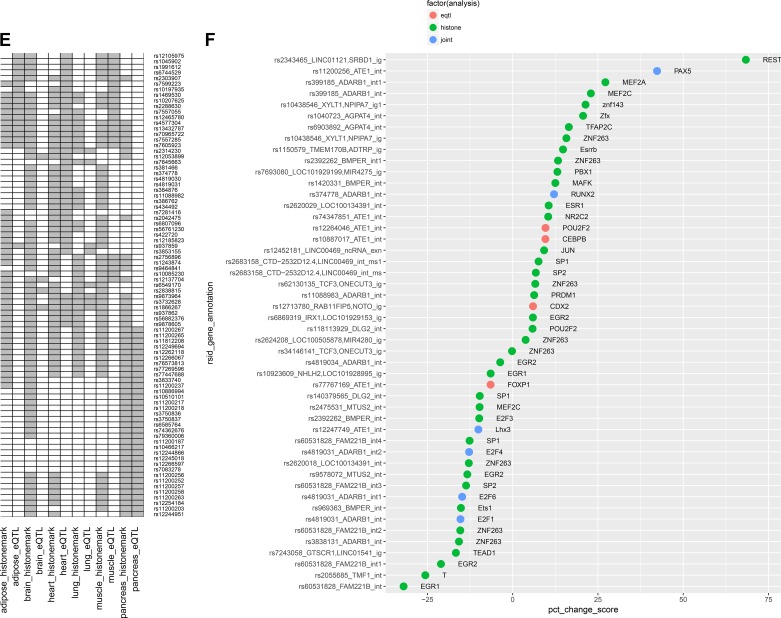
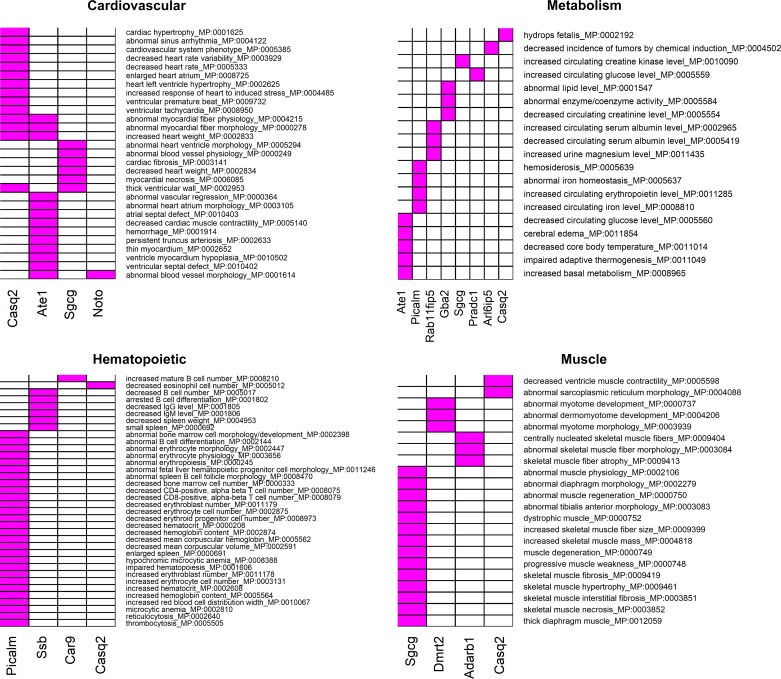
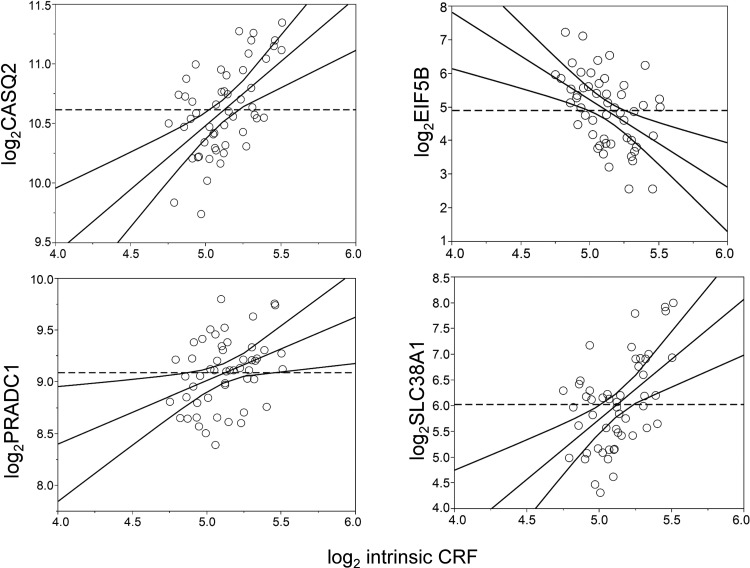
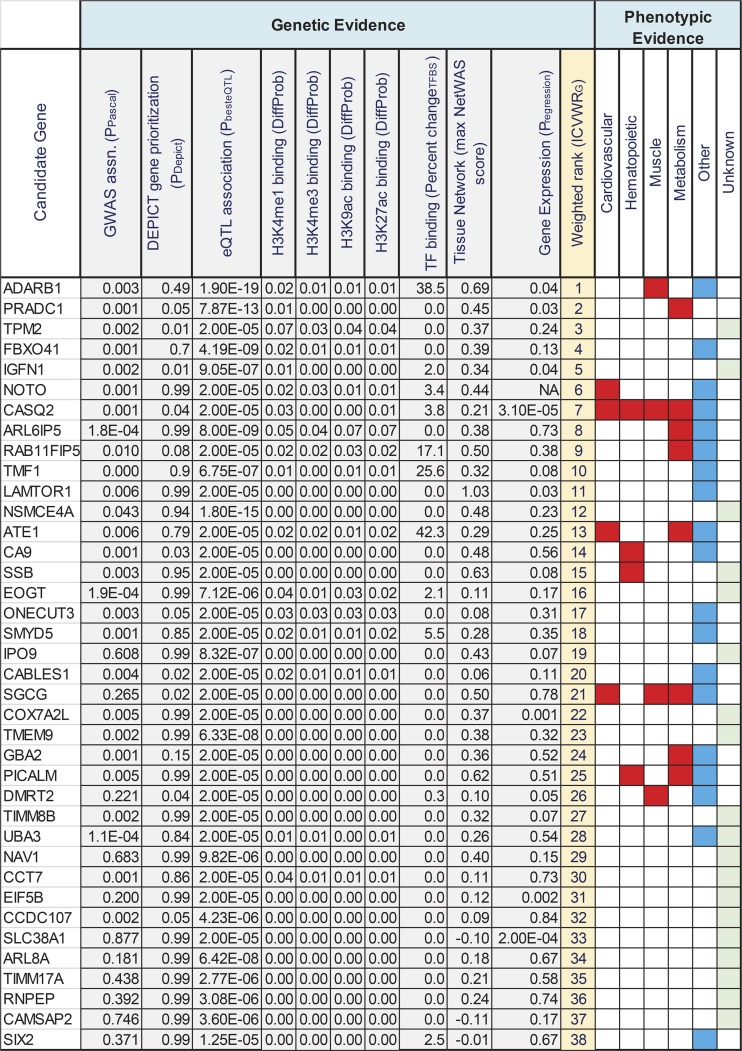
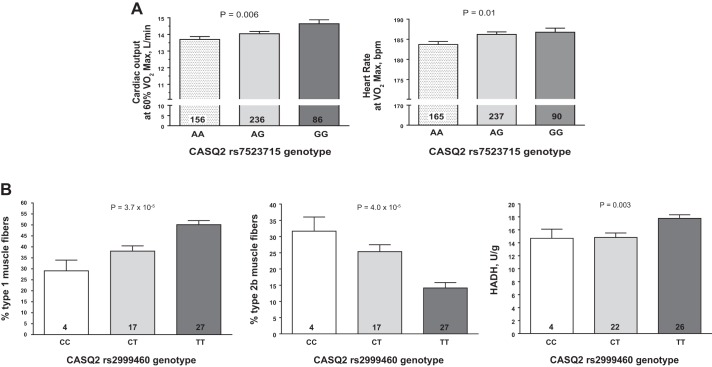
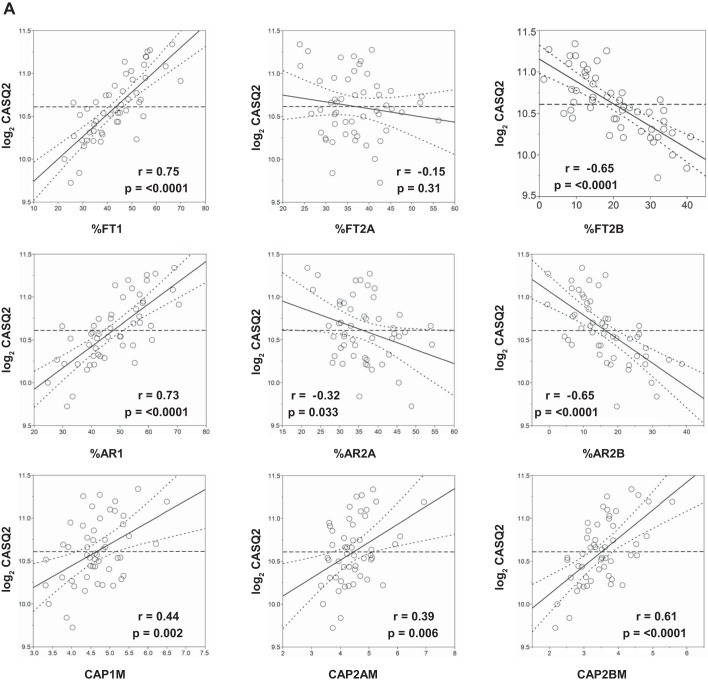
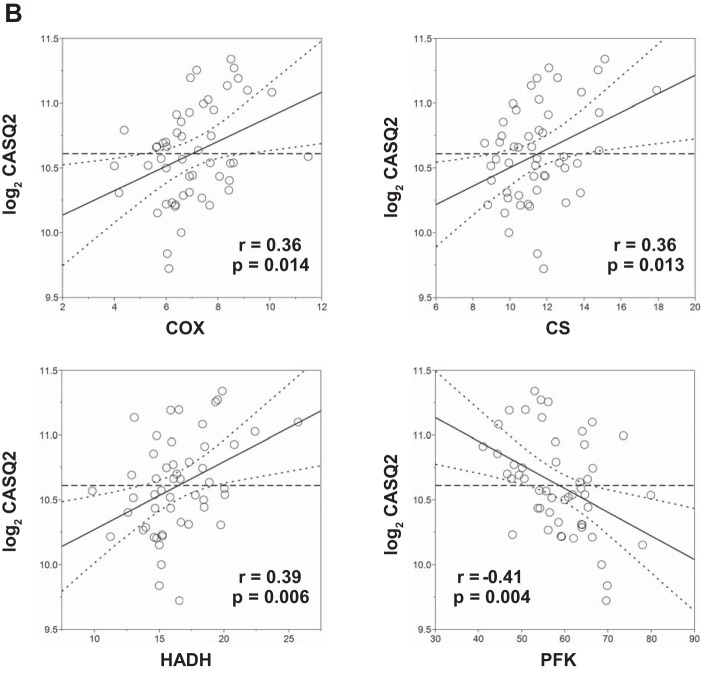
Intrinsic cardiorespiratory fitness (CRF) is defined as the level of CRF in the sedentary state. There are large individual differences in intrinsic CRF among sedentary adults. The physiology of variability in CRF has received much attention, but little is known about the genetic and molecular mechanisms that impact intrinsic CRF. These issues were explored in the present study by interrogating intrinsic CRF-associated DNA sequence variation and skeletal muscle gene expression data from the HERITAGE Family Study through an integrative bioinformatics guided approach. A combined analytic strategy involving genetic association, pathway enrichment, tissue-specific network structure, cis-regulatory genome effects, and expression quantitative trait loci was used to select and rank genes through a variation-adjusted weighted ranking scheme. Prioritized genes were further interrogated for corroborative evidence from knockout mouse phenotypes and relevant physiological traits from the HERITAGE cohort. The mean intrinsic V̇o2max was 33.1 ml O2·kg-1·min-1 (SD = 8.8) for the sample of 493 sedentary adults. Suggestive evidence was found for gene loci related to cardiovascular physiology (ATE1, CASQ2, NOTO, and SGCG), hematopoiesis (PICALM, SSB, CA9, and CASQ2), skeletal muscle phenotypes (SGCG, DMRT2, ADARB1, and CASQ2), and metabolism (ATE1, PICALM, RAB11FIP5, GBA2, SGCG, PRADC1, ARL6IP5, and CASQ2). Supportive evidence for a role of several of these loci was uncovered via association between DNA variants and muscle gene expression levels with exercise cardiovascular and muscle physiological traits. This initial effort to define the underlying molecular substrates of intrinsic CRF warrants further studies based on appropriate cohorts and study designs, complemented by functional investigations. NEW & NOTEWORTHY Intrinsic cardiorespiratory fitness (CRF) is measured in the sedentary state and is highly variable among sedentary adults. The physiology of variability in intrinsic cardiorespiratory fitness has received much attention, but little is known about the genetic and molecular mechanisms that impact intrinsic CRF. These issues were explored computationally in the present study, with further corroborative evidence obtained from analysis of phenotype data from knockout mouse models and human cardiovascular and skeletal muscle measurements.
Keywords: bioinformatics; cardiovascular physiology; in silico exploration of the biology of cardiorespiratory fitness; intrinsic cardiorespiratory fitness; skeletal muscle biology.
Conflict of interest statement
M.A. Sarzynski is a consultant for Genetic Direction. The other authors have no conflicts of interest to declare.
Figures

Similar articles
-
The combination of cardiorespiratory fitness and muscle strength, and mortality risk.Eur J Epidemiol. 2018 Oct;33(10):953-964. doi: 10.1007/s10654-018-0384-x. Epub 2018 Mar 28. Eur J Epidemiol. 2018. PMID: 29594847 Free PMC article.
-
The genetic case for cardiorespiratory fitness as a clinical vital sign and the routine prescription of physical activity in healthcare.Genome Med. 2021 Nov 9;13(1):180. doi: 10.1186/s13073-021-00994-9. Genome Med. 2021. PMID: 34753499 Free PMC article.
-
Omics-driven investigation of the biology underlying intrinsic submaximal working capacity and its trainability.Physiol Genomics. 2023 Nov 1;55(11):517-543. doi: 10.1152/physiolgenomics.00163.2022. Epub 2023 Sep 4. Physiol Genomics. 2023. PMID: 37661925 Free PMC article.
-
Epidemiological, mechanistic, and practical bases for assessment of cardiorespiratory fitness and muscle status in adults in healthcare settings.Eur J Appl Physiol. 2023 May;123(5):945-964. doi: 10.1007/s00421-022-05114-y. Epub 2023 Jan 23. Eur J Appl Physiol. 2023. PMID: 36683091 Free PMC article. Review.
-
Effects of cardiorespiratory fitness and exercise training on cerebrovascular blood flow and reactivity: a systematic review with meta-analyses.Am J Physiol Heart Circ Physiol. 2021 Jul 1;321(1):H59-H76. doi: 10.1152/ajpheart.00880.2020. Epub 2021 May 21. Am J Physiol Heart Circ Physiol. 2021. PMID: 34018848
Cited by
-
Understanding the Relationship between Intrinsic Cardiorespiratory Fitness and Serum and Skeletal Muscle Metabolomics Profile.J Proteome Res. 2021 May 7;20(5):2397-2409. doi: 10.1021/acs.jproteome.0c00905. Epub 2021 Apr 28. J Proteome Res. 2021. PMID: 33909435 Free PMC article.
-
DMRT2 Interacts With FXR and Improves Insulin Resistance in Adipocytes and a Mouse Model.Front Endocrinol (Lausanne). 2022 Feb 17;12:723623. doi: 10.3389/fendo.2021.723623. eCollection 2021. Front Endocrinol (Lausanne). 2022. PMID: 35250844 Free PMC article.
-
Circular RNA Involvement in the Protective Effect of Human Umbilical Cord Mesenchymal Stromal Cell-Derived Extracellular Vesicles Against Hypoxia/Reoxygenation Injury in Cardiac Cells.Front Cardiovasc Med. 2021 Feb 23;8:626878. doi: 10.3389/fcvm.2021.626878. eCollection 2021. Front Cardiovasc Med. 2021. PMID: 33708804 Free PMC article.
-
Sex-specific Genetic Determinants of Right Ventricular Structure and Function.Am J Respir Crit Care Med. 2024 Oct 7;211(1):113-23. doi: 10.1164/rccm.202404-0721OC. Online ahead of print. Am J Respir Crit Care Med. 2024. PMID: 39374572 Free PMC article.
-
World-class athletic performance and genetic endowment.Nat Metab. 2020 Sep;2(9):796-798. doi: 10.1038/s42255-020-0233-6. Nat Metab. 2020. PMID: 32943784 Review. No abstract available.
References
-
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, Farnham PJ, Hirst M, Lander ES, Mikkelsen TS, Thomson JA. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol 28: 1045–1048, 2010. doi:10.1038/nbt1010-1045. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous