

Bias-minimized quantification of microRNA reveals widespread alternative processing and 3' end modification
- PMID: 30605524
- PMCID: PMC6411932
- DOI: 10.1093/nar/gky1293
Bias-minimized quantification of microRNA reveals widespread alternative processing and 3' end modification
Erratum in
-
Correction to 'Bias-minimized quantification of microRNA reveals widespread alternative processing and 3' end modification'.Nucleic Acids Res. 2022 Feb 22;50(3):1799-1800. doi: 10.1093/nar/gkac078. Nucleic Acids Res. 2022. PMID: 35104843 Free PMC article. No abstract available.
Abstract
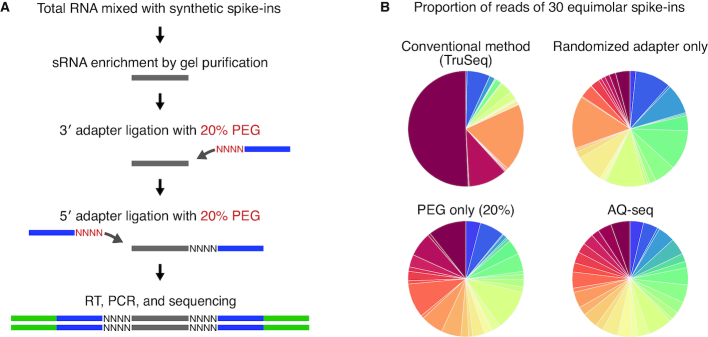
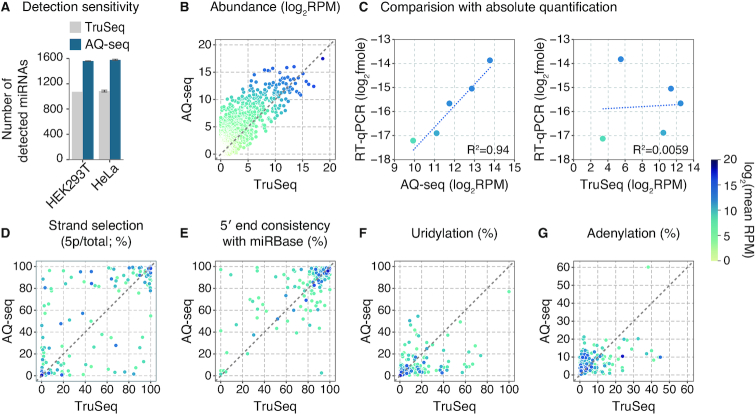
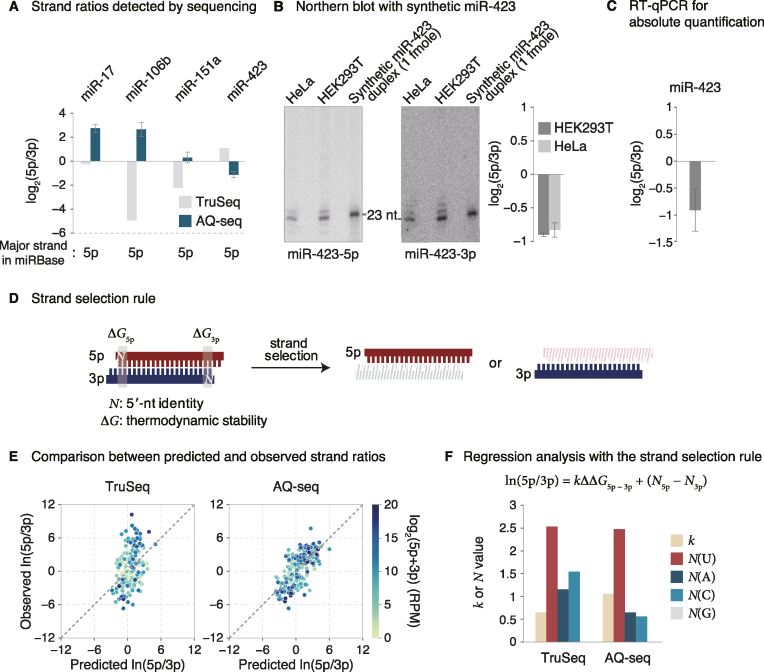
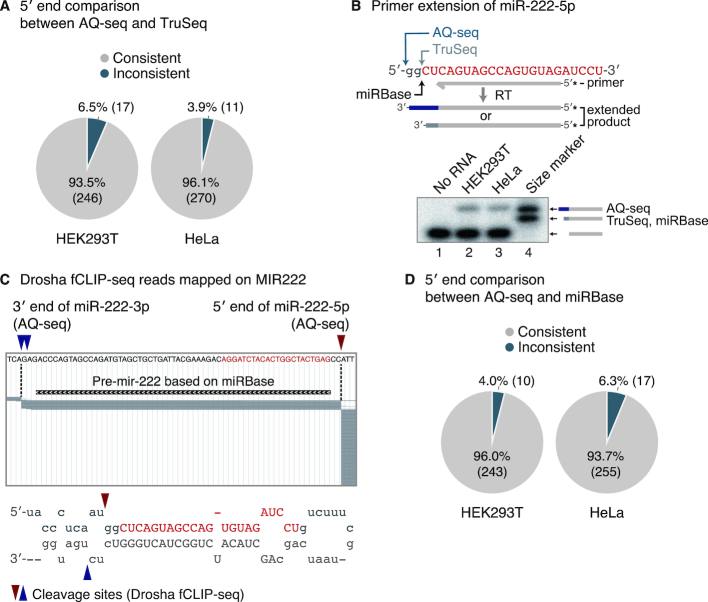
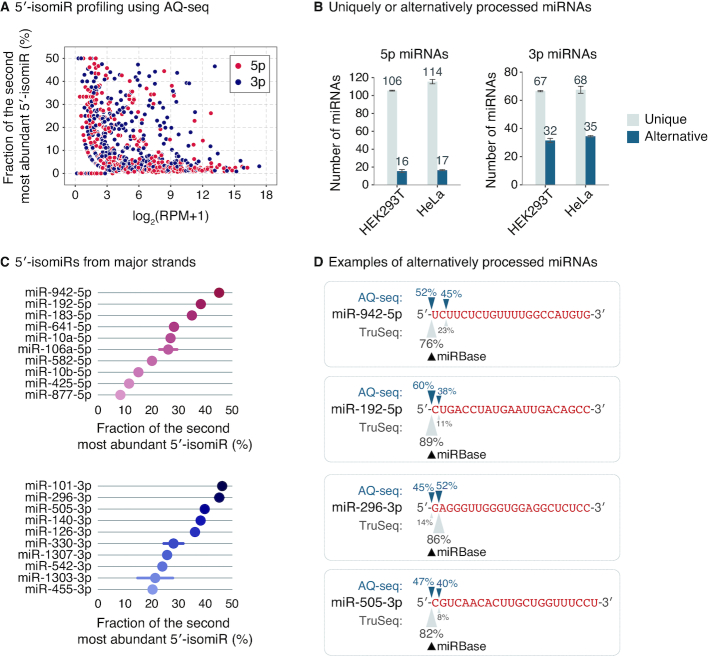
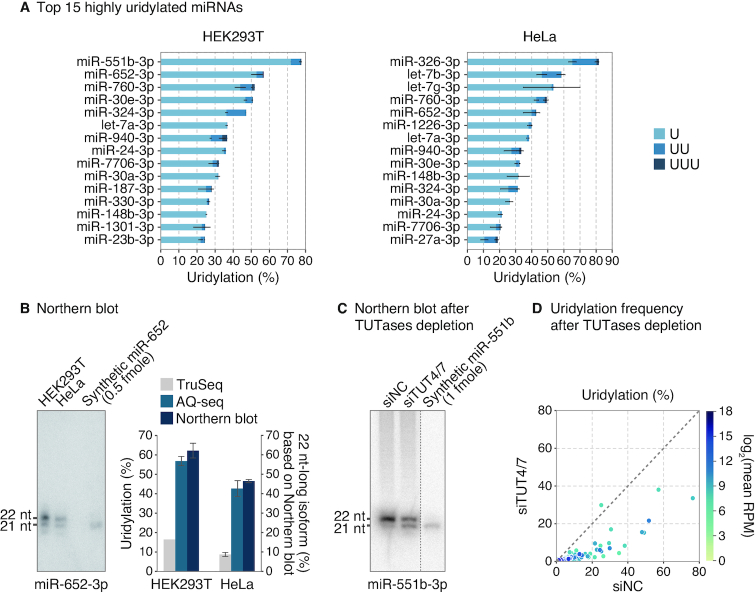
MicroRNAs (miRNAs) modulate diverse biological and pathological processes via post-transcriptional gene silencing. High-throughput small RNA sequencing (sRNA-seq) has been widely adopted to investigate the functions and regulatory mechanisms of miRNAs. However, accurate quantification of miRNAs has been limited owing to the severe ligation bias in conventional sRNA-seq methods. Here, we quantify miRNAs and their variants (known as isomiRs) by an improved sRNA-seq protocol, termed AQ-seq (accurate quantification by sequencing), that utilizes adapters with terminal degenerate sequences and a high concentration of polyethylene glycol (PEG), which minimize the ligation bias during library preparation. Measurement using AQ-seq allows us to correct the previously misannotated 5' end usage and strand preference in public databases. Importantly, the analysis of 5' terminal heterogeneity reveals widespread alternative processing events which have been underestimated. We also identify highly uridylated miRNAs originating from the 3p strands, indicating regulations mediated by terminal uridylyl transferases at the pre-miRNA stage. Taken together, our study reveals the complexity of the miRNA isoform landscape, allowing us to refine miRNA annotation and to advance our understanding of miRNA regulation. Furthermore, AQ-seq can be adopted to improve other ligation-based sequencing methods including crosslinking-immunoprecipitation-sequencing (CLIP-seq) and ribosome profiling (Ribo-seq).
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Ha M., Kim V.N.. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014; 15:509–524. - PubMed
-
- Lee Y., Ahn C., Han J., Choi H., Kim J., Yim J., Lee J., Provost P., Radmark O., Kim S. et al. .. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003; 425:415–419. - PubMed
-
- Denli A.M., Tops B.B., Plasterk R.H., Ketting R.F., Hannon G.J.. Processing of primary microRNAs by the microprocessor complex. Nature. 2004; 432:231–235. - PubMed
-
- Gregory R.I., Yan K.P., Amuthan G., Chendrimada T., Doratotaj B., Cooch N., Shiekhattar R.. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004; 432:235–240. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
