

An AR-ERG transcriptional signature defined by long-range chromatin interactomes in prostate cancer cells
- PMID: 30606742
- PMCID: PMC6360806
- DOI: 10.1101/gr.230243.117
An AR-ERG transcriptional signature defined by long-range chromatin interactomes in prostate cancer cells
Abstract
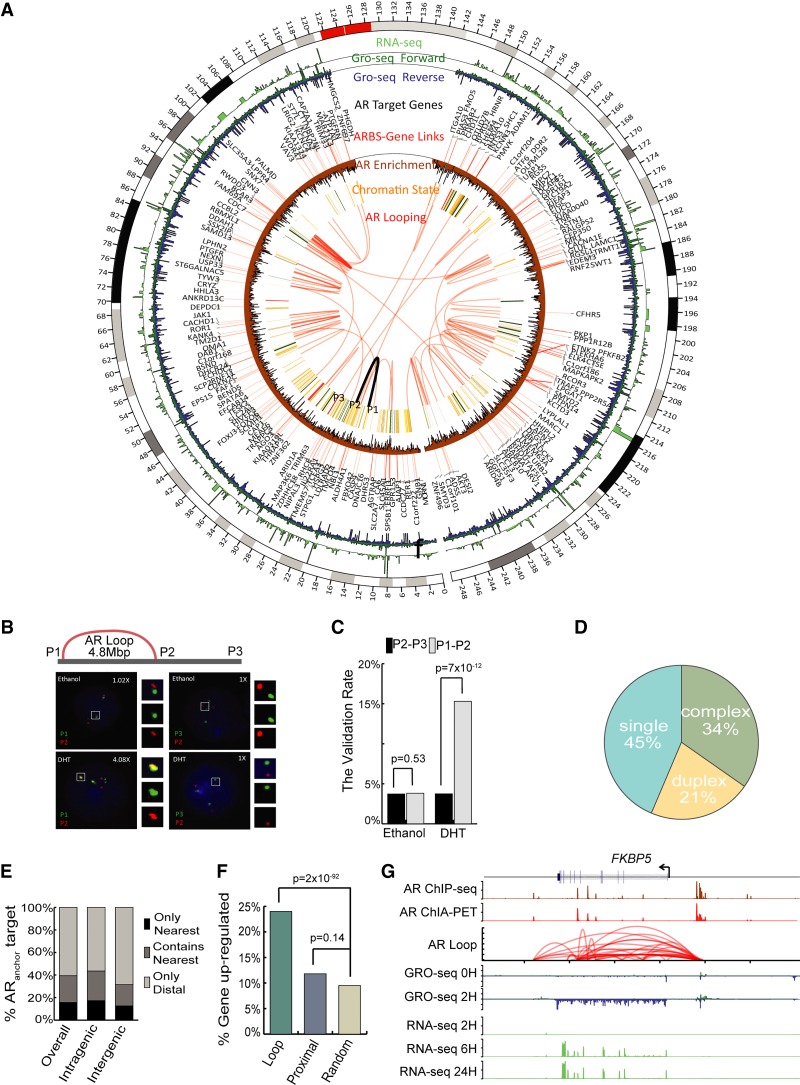
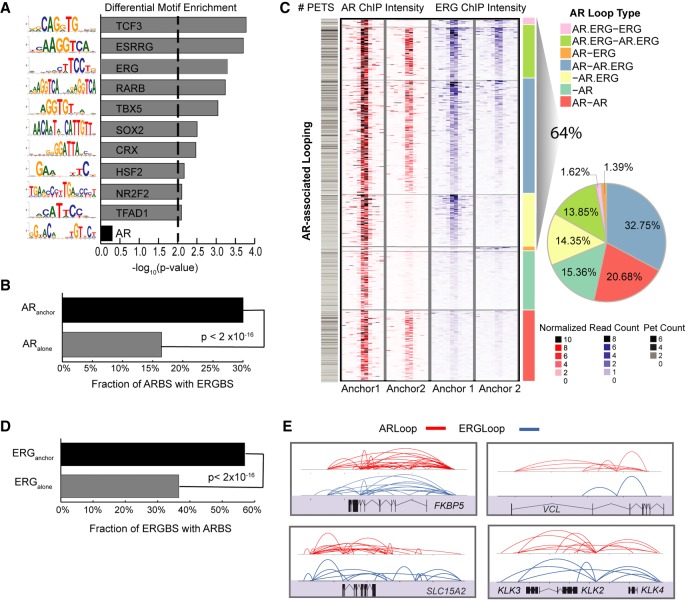
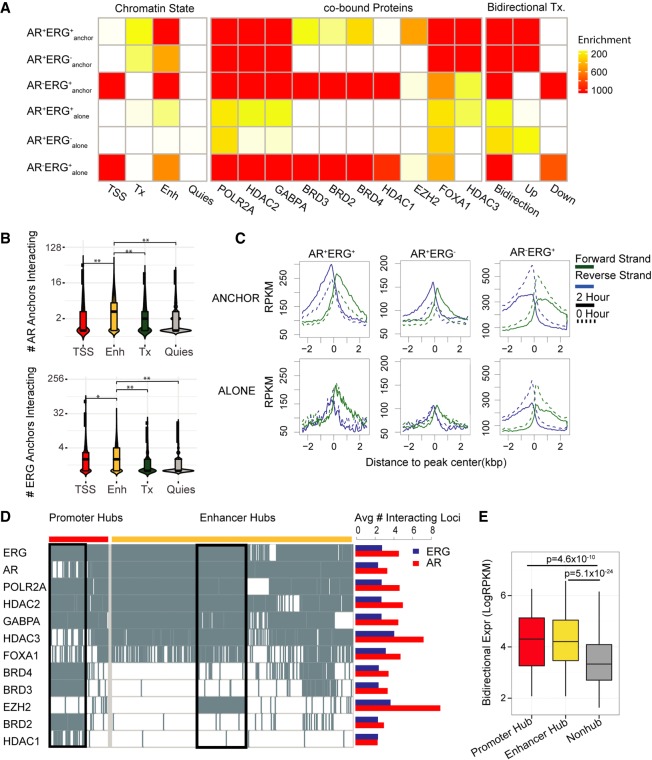
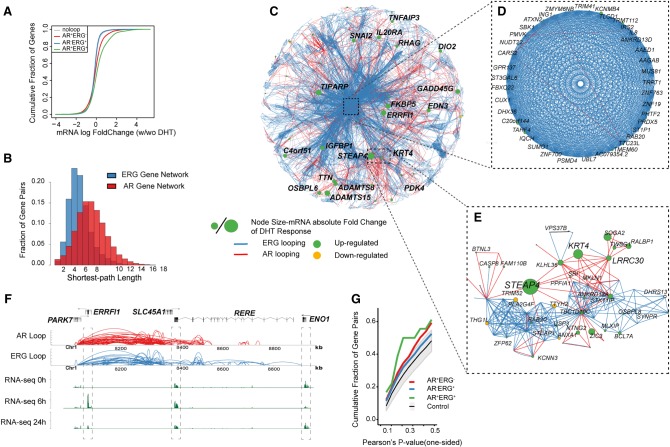
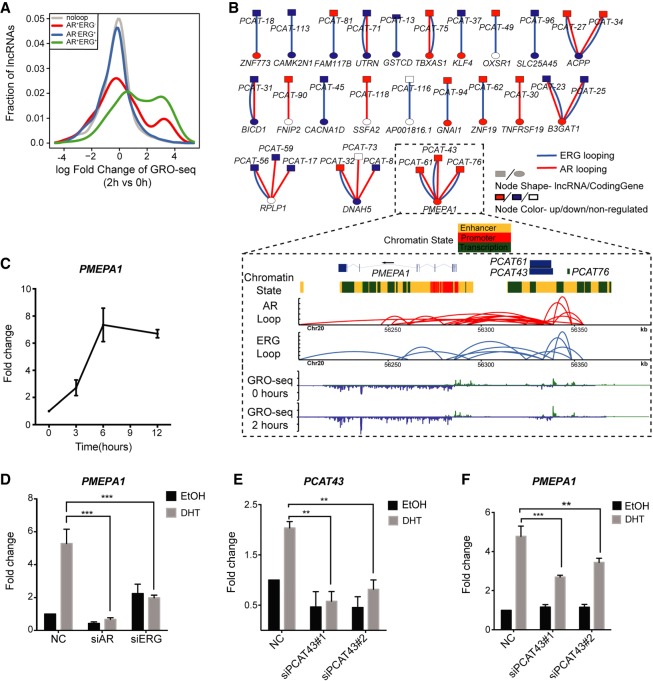
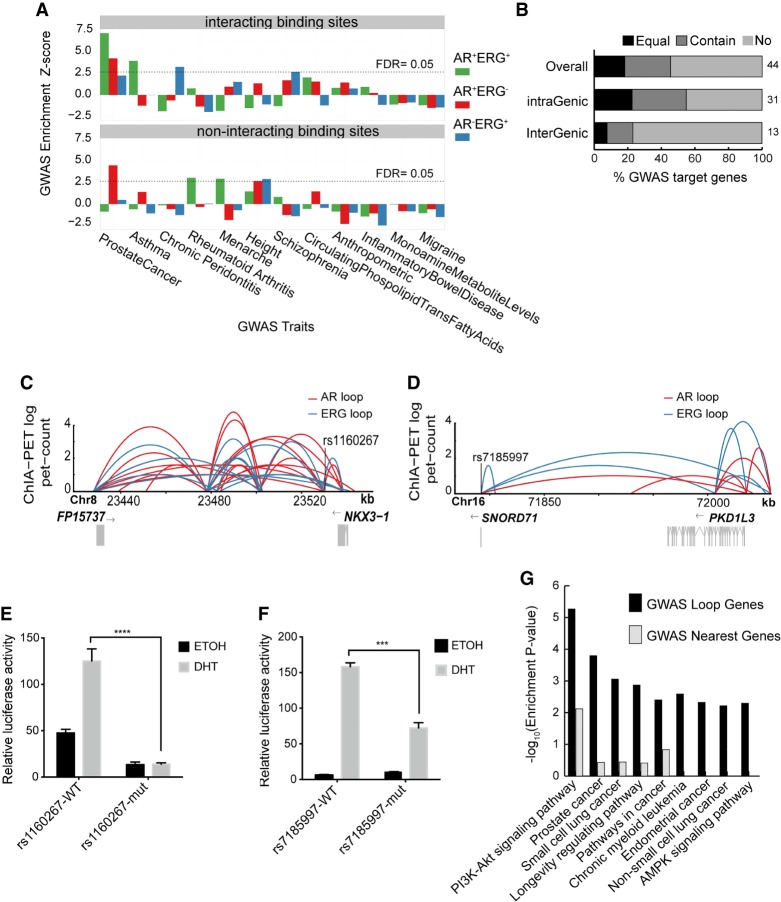
The aberrant activities of transcription factors such as the androgen receptor (AR) underpin prostate cancer development. While the AR cis-regulation has been extensively studied in prostate cancer, information pertaining to the spatial architecture of the AR transcriptional circuitry remains limited. In this paper, we propose a novel framework to profile long-range chromatin interactions associated with AR and its collaborative transcription factor, erythroblast transformation-specific related gene (ERG), using chromatin interaction analysis by paired-end tag (ChIA-PET). We identified ERG-associated long-range chromatin interactions as a cooperative component in the AR-associated chromatin interactome, acting in concert to achieve coordinated regulation of a subset of AR target genes. Through multifaceted functional data analysis, we found that AR-ERG interaction hub regions are characterized by distinct functional signatures, including bidirectional transcription and cotranscription factor binding. In addition, cancer-associated long noncoding RNAs were found to be connected near protein-coding genes through AR-ERG looping. Finally, we found strong enrichment of prostate cancer genome-wide association study (GWAS) single nucleotide polymorphisms (SNPs) at AR-ERG co-binding sites participating in chromatin interactions and gene regulation, suggesting GWAS target genes identified from chromatin looping data provide more biologically relevant findings than using the nearest gene approach. Taken together, our results revealed an AR-ERG-centric higher-order chromatin structure that drives coordinated gene expression in prostate cancer progression and the identification of potential target genes for therapeutic intervention.
© 2019 Zhang et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials