

Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias
- PMID: 30607747
- PMCID: PMC6554265
- DOI: 10.1007/s13311-018-00696-y
Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias
Abstract
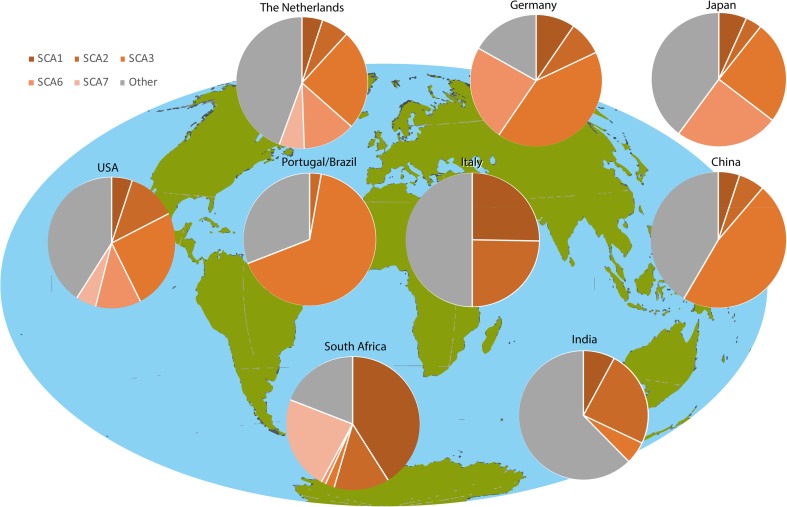
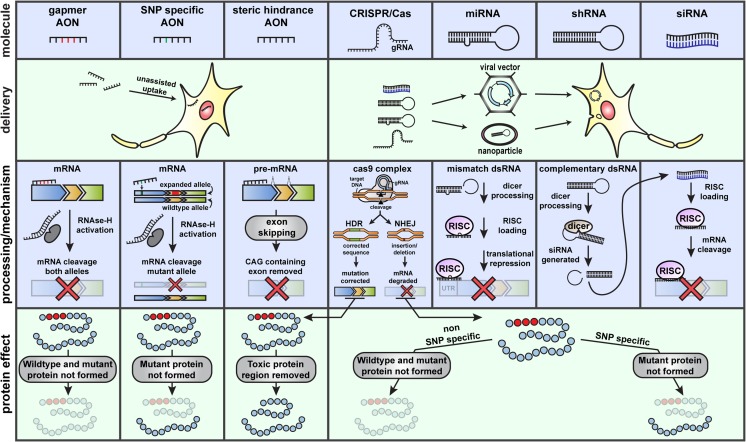
Autosomal dominant cerebellar ataxias (ADCAs) are a group of neurodegenerative disorders characterized by degeneration of the cerebellum and its connections. All ADCAs have progressive ataxia as their main clinical feature, frequently accompanied by dysarthria and oculomotor deficits. The most common spinocerebellar ataxias (SCAs) are 6 polyglutamine (polyQ) SCAs. These diseases are all caused by a CAG repeat expansion in the coding region of a gene. Currently, no curative treatment is available for any of the polyQ SCAs, but increasing knowledge on the genetics and the pathological mechanisms of these polyQ SCAs has provided promising therapeutic targets to potentially slow disease progression. Potential treatments can be divided into pharmacological and gene therapies that target the toxic downstream effects, gene therapies that target the polyQ SCA genes, and stem cell replacement therapies. Here, we will provide a review on the genetics, mechanisms, and therapeutic progress in polyglutamine spinocerebellar ataxias.
Keywords: SCA; Spinocerebellar ataxia; antisense oligonucleotides; gene therapy; polyglutamine disorders; stem cell-based therapy.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
