

Folding and Misfolding of Human Membrane Proteins in Health and Disease: From Single Molecules to Cellular Proteostasis
- PMID: 30608666
- PMCID: PMC6506414
- DOI: 10.1021/acs.chemrev.8b00532
Folding and Misfolding of Human Membrane Proteins in Health and Disease: From Single Molecules to Cellular Proteostasis
Abstract
Advances over the past 25 years have revealed much about how the structural properties of membranes and associated proteins are linked to the thermodynamics and kinetics of membrane protein (MP) folding. At the same time biochemical progress has outlined how cellular proteostasis networks mediate MP folding and manage misfolding in the cell. When combined with results from genomic sequencing, these studies have established paradigms for how MP folding and misfolding are linked to the molecular etiologies of a variety of diseases. This emerging framework has paved the way for the development of a new class of small molecule "pharmacological chaperones" that bind to and stabilize misfolded MP variants, some of which are now in clinical use. In this review, we comprehensively outline current perspectives on the folding and misfolding of integral MPs as well as the mechanisms of cellular MP quality control. Based on these perspectives, we highlight new opportunities for innovations that bridge our molecular understanding of the energetics of MP folding with the nuanced complexity of biological systems. Given the many linkages between MP misfolding and human disease, we also examine some of the exciting opportunities to leverage these advances to address emerging challenges in the development of therapeutics and precision medicine.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
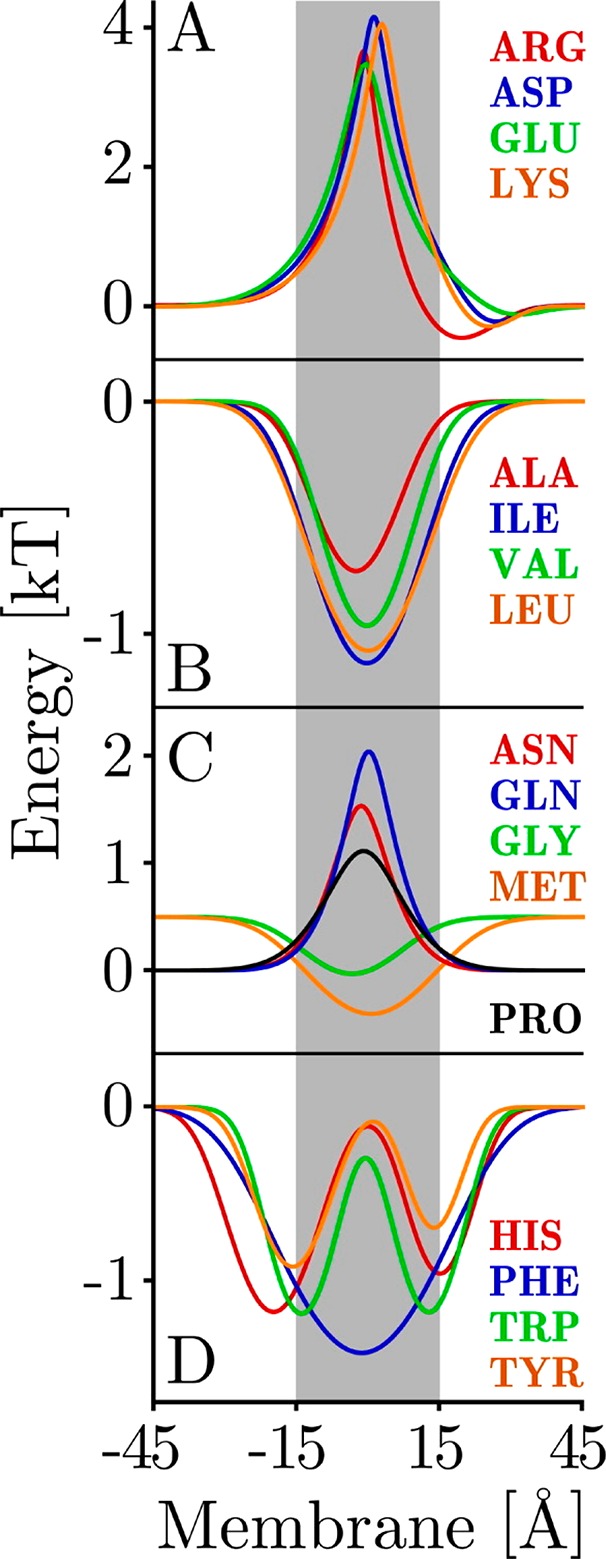
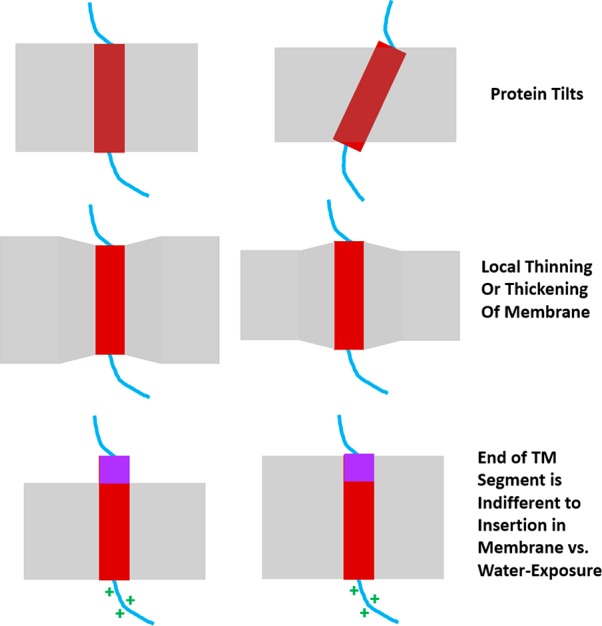
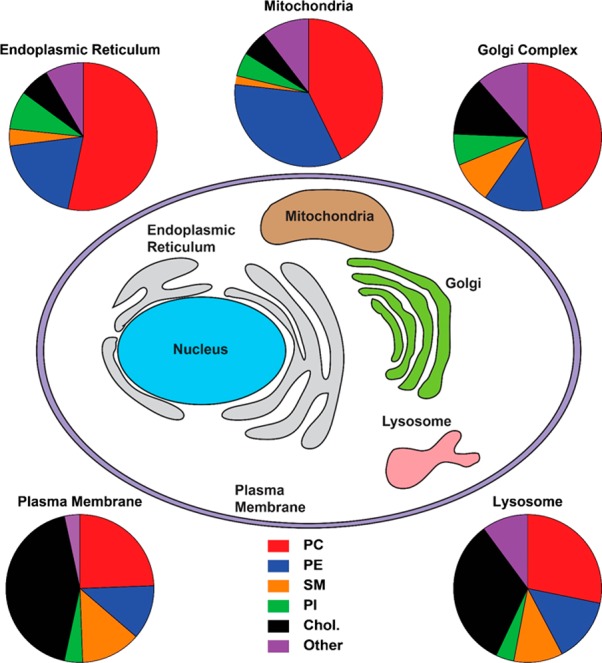
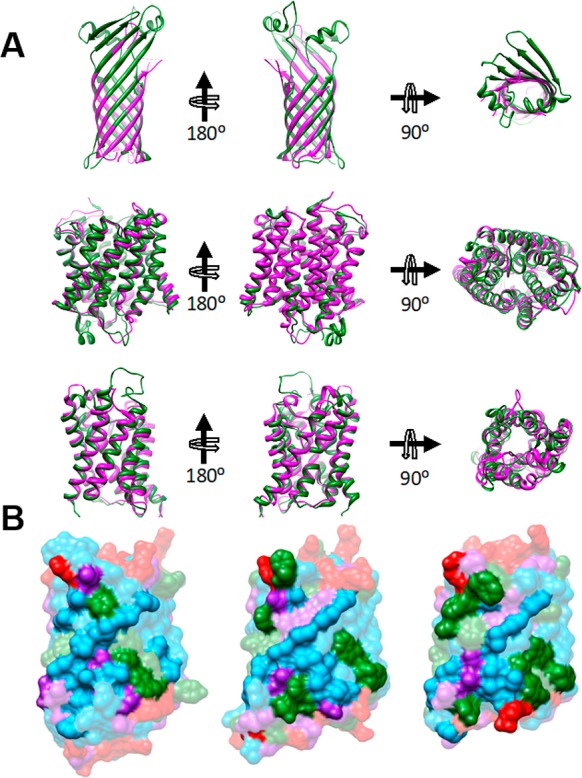
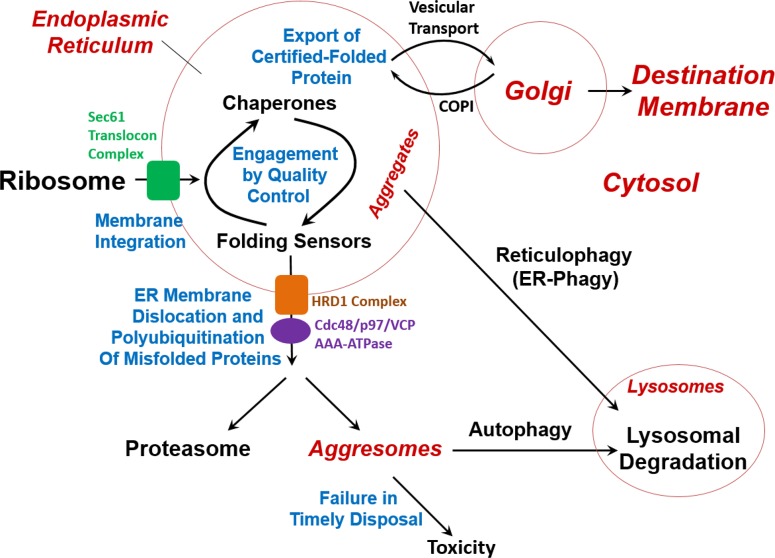
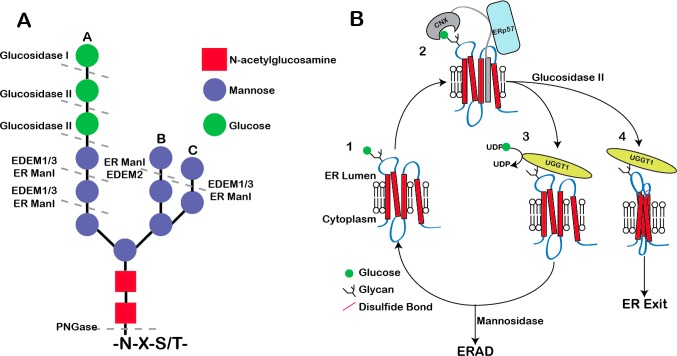
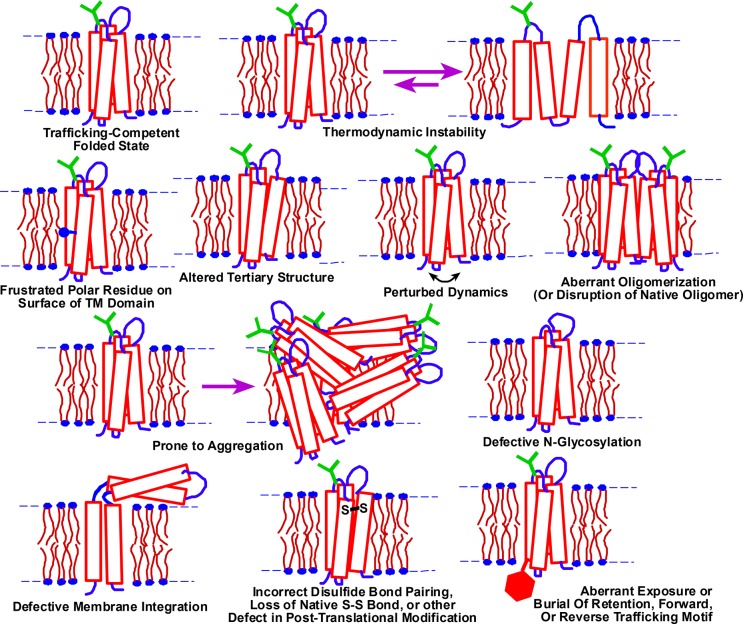
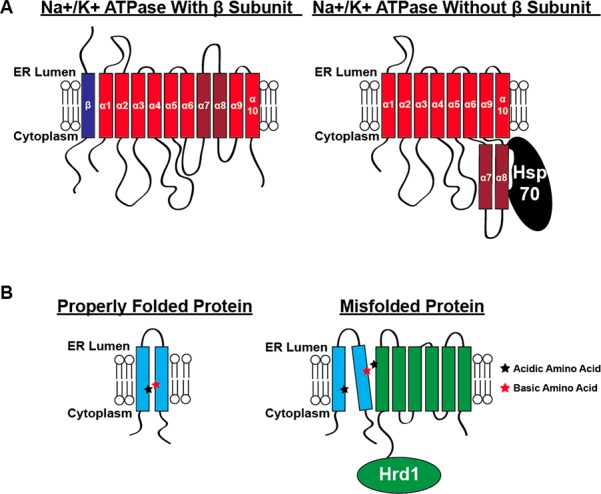
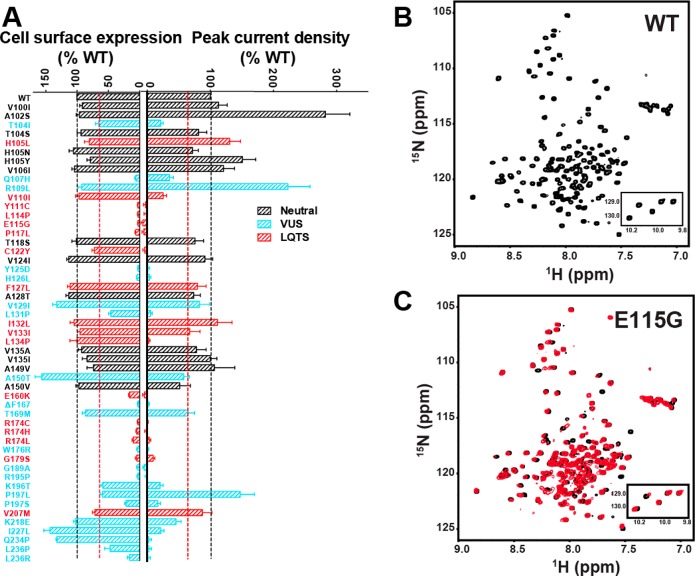
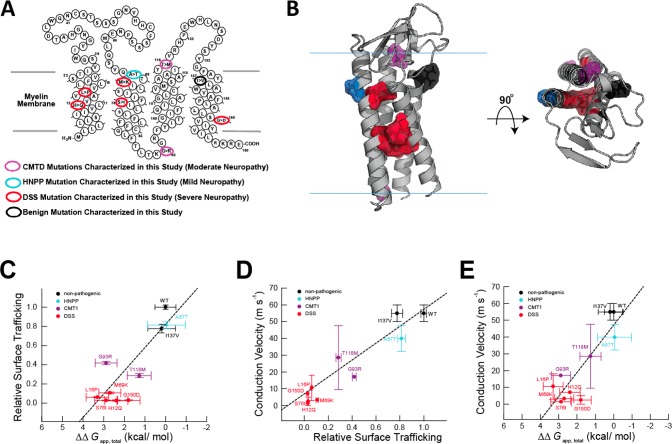
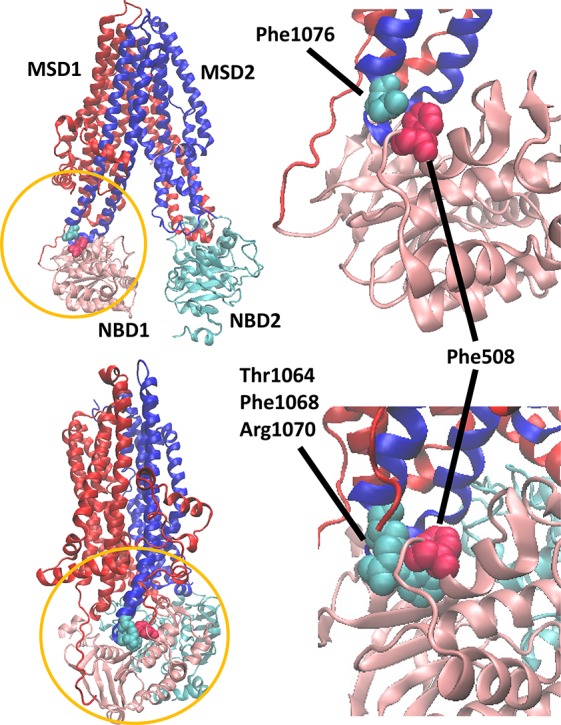
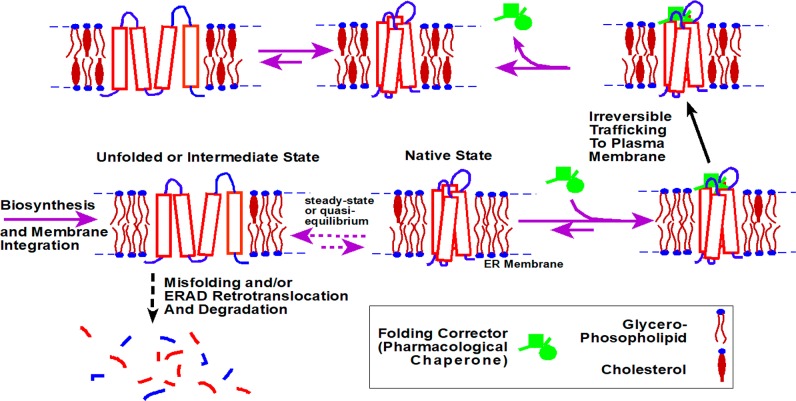
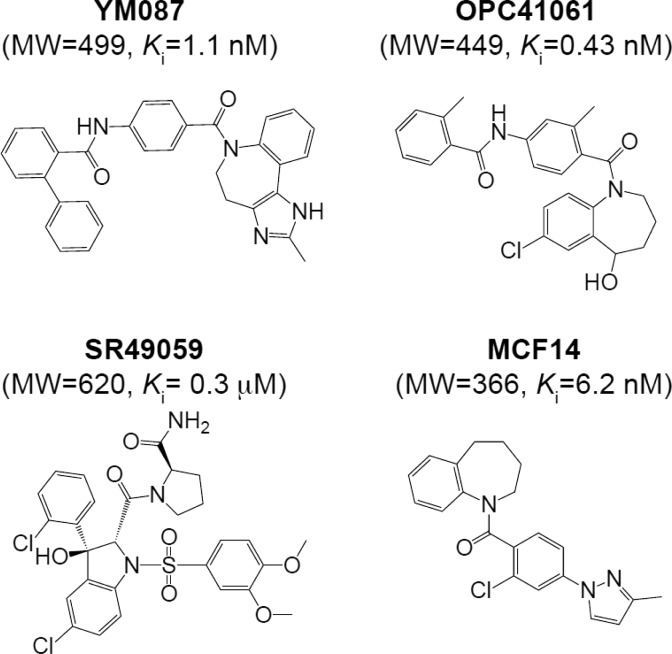
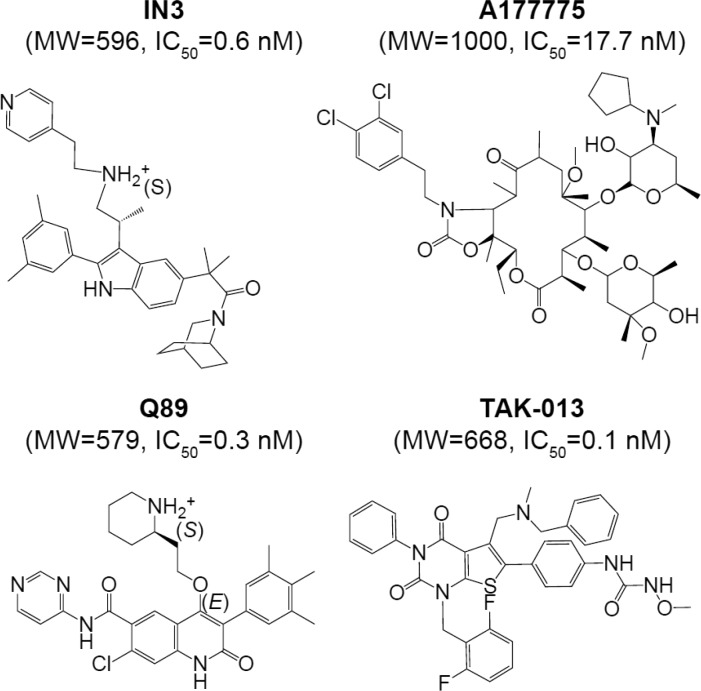
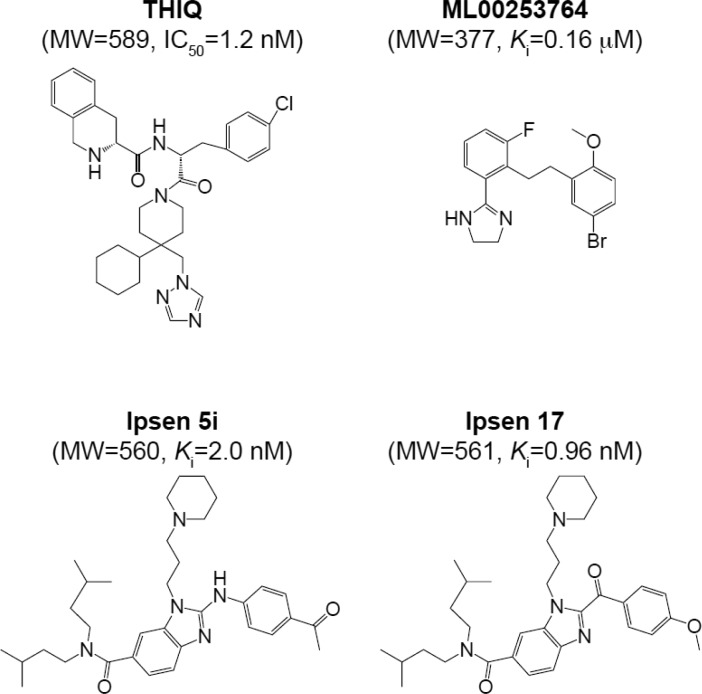
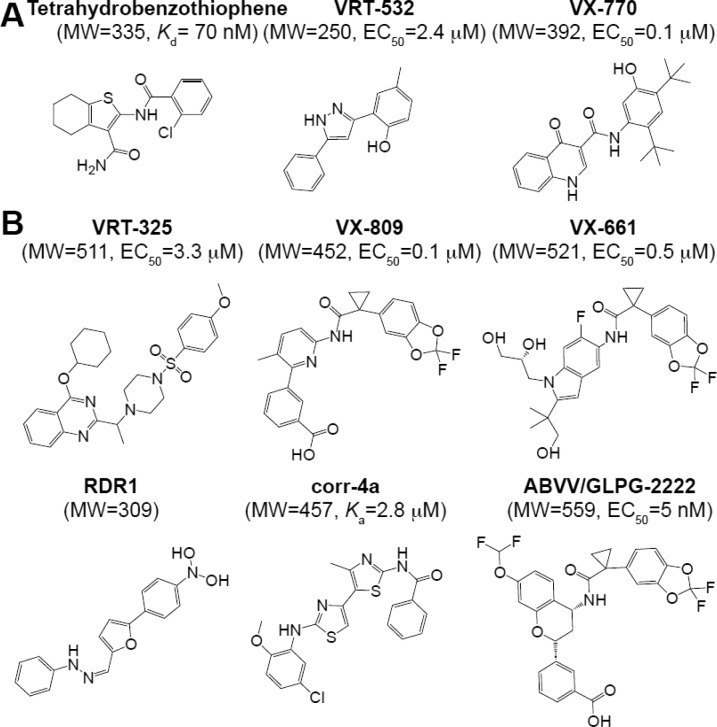
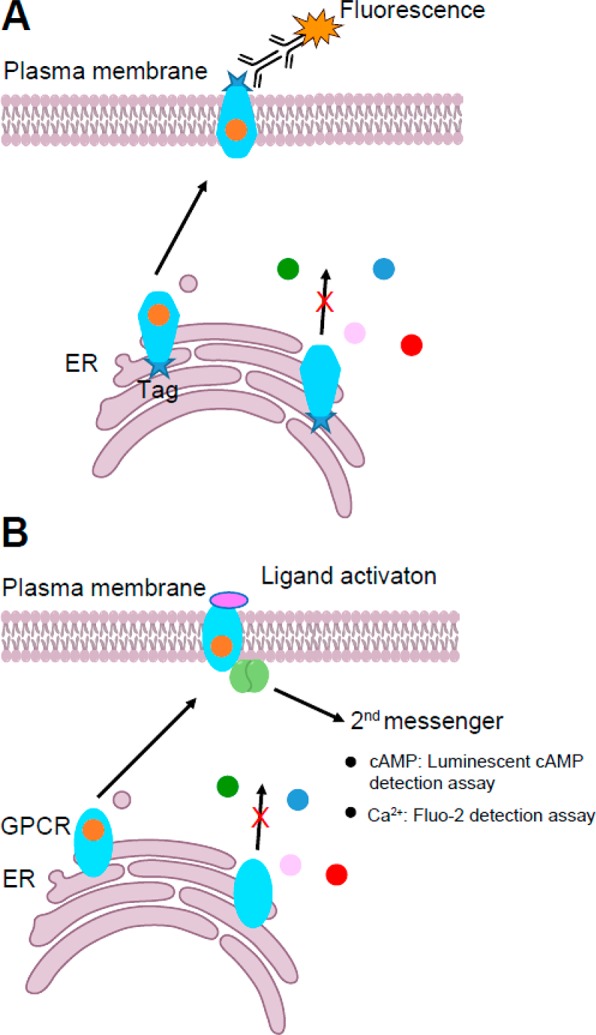
References
-
- Huang K. S.; Bayley H.; Liao M. J.; London E.; Khorana H. G. Refolding of an integral membrane protein. Denaturation, renaturation, and reconstitution of intact bacteriorhodopsin and two proteolytic fragments. J. Biol. Chem. 1981, 256, 3802–3809. - PubMed
-
- London E.; Khorana H. G. Denaturation and renaturation of bacteriorhodopsin in detergents and lipid-detergent mixtures. J. Biol. Chem. 1982, 257, 7003–7011. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
