

Niche-directed evolution modulates genome architecture in freshwater Planctomycetes
- PMID: 30610231
- PMCID: PMC6461901
- DOI: 10.1038/s41396-018-0332-5
Niche-directed evolution modulates genome architecture in freshwater Planctomycetes
Abstract
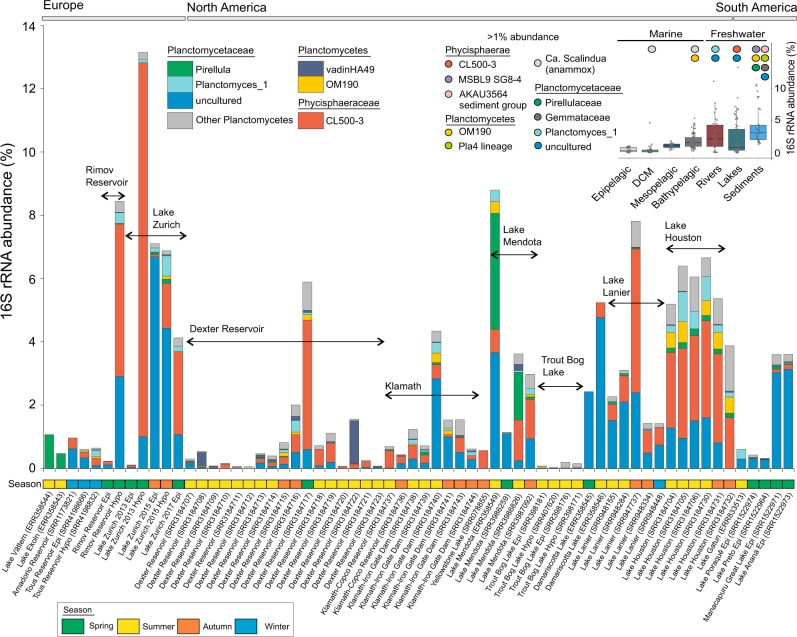
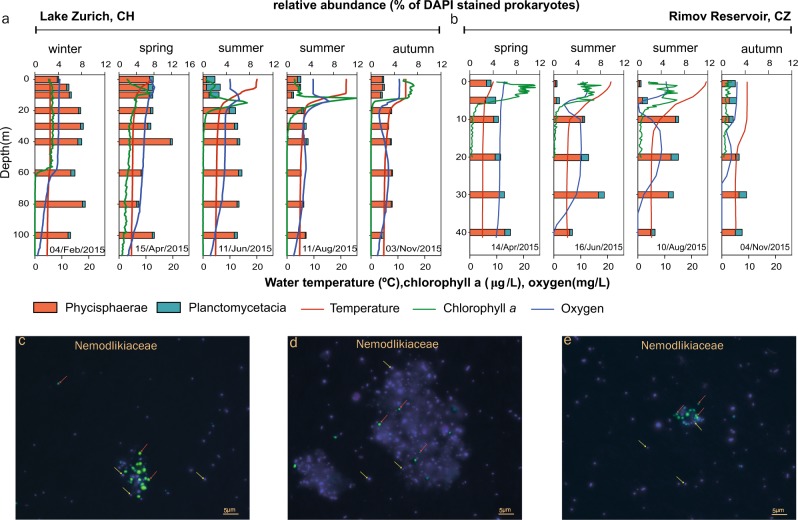
Freshwater environments teem with microbes that do not have counterparts in culture collections or genetic data available in genomic repositories. Currently, our apprehension of evolutionary ecology of freshwater bacteria is hampered by the difficulty to establish organism models for the most representative clades. To circumvent the bottlenecks inherent to the cultivation-based techniques, we applied ecogenomics approaches in order to unravel the evolutionary history and the processes that drive genome architecture in hallmark freshwater lineages from the phylum Planctomycetes. The evolutionary history inferences showed that sediment/soil Planctomycetes transitioned to aquatic environments, where they gave rise to new freshwater-specific clades. The most abundant lineage was found to have the most specialised lifestyle (increased regulatory genetic circuits, metabolism tuned for mineralization of proteinaceous sinking aggregates, psychrotrophic behaviour) within the analysed clades and to harbour the smallest freshwater Planctomycetes genomes, highlighting a genomic architecture shaped by niche-directed evolution (through loss of functions and pathways not needed in the newly acquired freshwater niche).
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures



References
-
- Woese CR, Stackebrandt E, Macke TJ, Fox GE. A phylogenetic definition of the major eubacterial taxa. Syst Appl Microbiol. 1985;6:143–51. - PubMed
-
- Staley JT. Budding bacteria of the Pasteuria–Blastobacter group. Can J Microbiol. 1973;19:609–14. - PubMed
-
- Garrity George M., Holt John G. Bergey’s Manual® of Systematic Bacteriology. New York, NY: Springer New York; 2001. The Road Map to the Manual; pp. 119–166.
-
- Erko S, Ludvig W, Schubert W, Klink F, Schlesner H, et al. Molecular genetic evidence for early evolutionary origin of budding peptidoglycan-less Eubacteria. Nature. 1984;307:735–7357. - PubMed
-
- Devos DP, Reynaud EG. Intermediate steps. Science (80-) 2010;330:1187–8. - PubMed
