

'Dusty core disease' (DuCD): expanding morphological spectrum of RYR1 recessive myopathies
- PMID: 30611313
- PMCID: PMC6320585
- DOI: 10.1186/s40478-018-0655-5
'Dusty core disease' (DuCD): expanding morphological spectrum of RYR1 recessive myopathies
Abstract
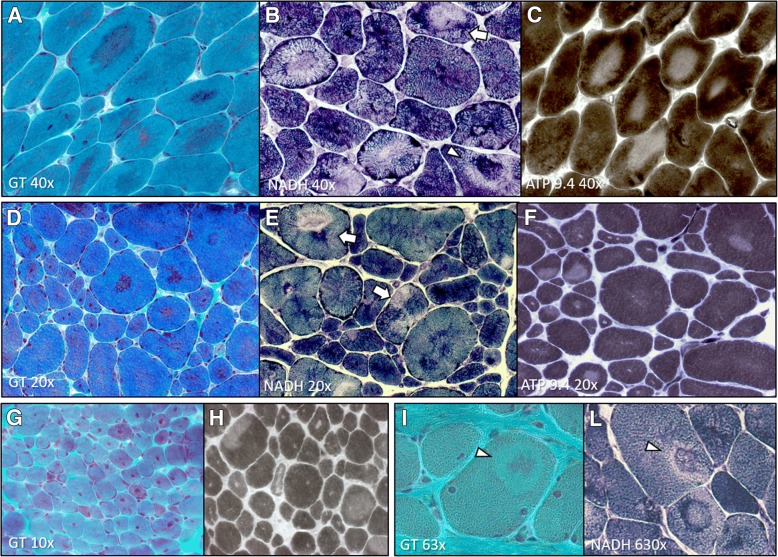
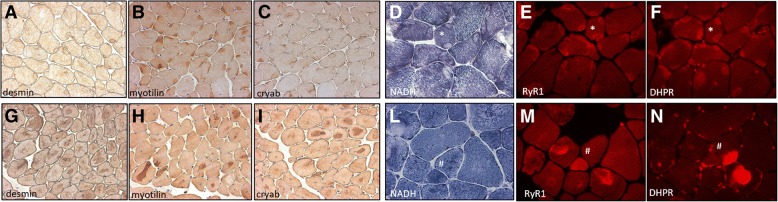
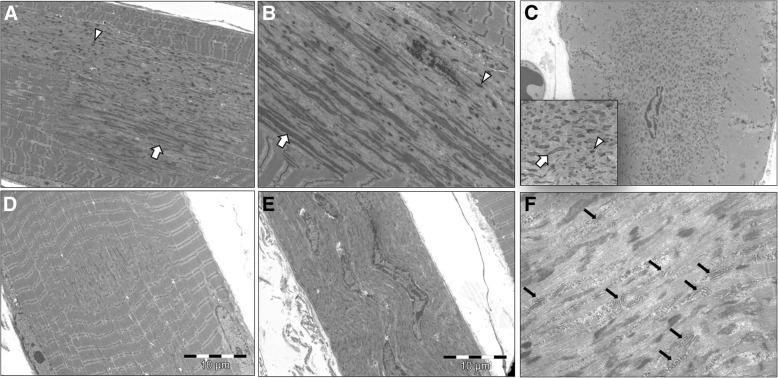
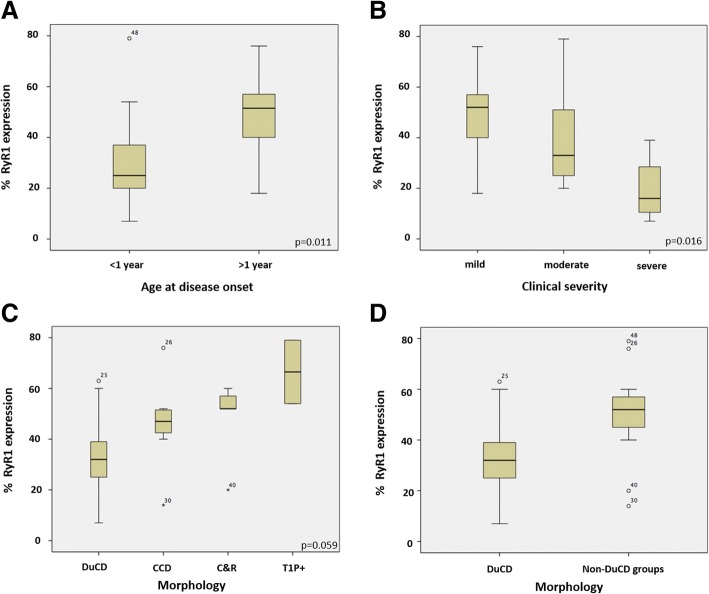
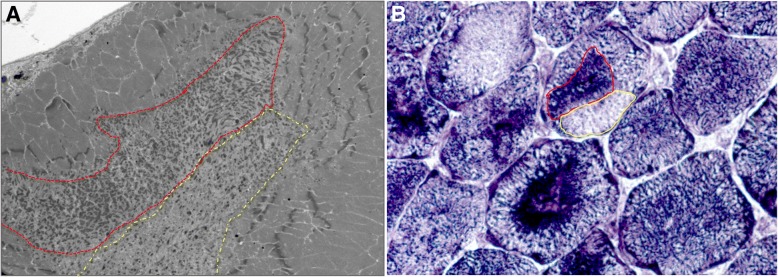
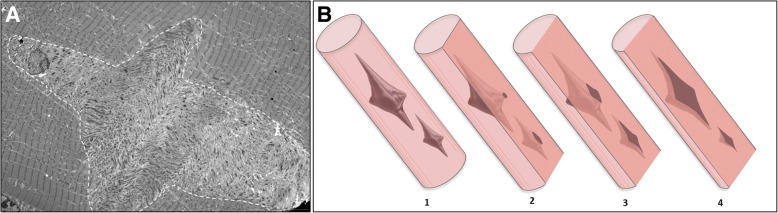
Several morphological phenotypes have been associated to RYR1-recessive myopathies. We recharacterized the RYR1-recessive morphological spectrum by a large monocentric study performed on 54 muscle biopsies from a large cohort of 48 genetically confirmed patients, using histoenzymology, immunohistochemistry, and ultrastructural studies. We also analysed the level of RyR1 expression in patients' muscle biopsies. We defined "dusty cores" the irregular areas of myofibrillar disorganisation characterised by a reddish-purple granular material deposition with uneven oxidative stain and devoid of ATPase activity, which represent the characteristic lesion in muscle biopsy in 54% of patients. We named Dusty Core Disease (DuCD) the corresponding entity of congenital myopathy. Dusty cores had peculiar histological and ultrastructural characteristics compared to the other core diseases. DuCD muscle biopsies also showed nuclear centralization and type1 fibre predominance. Dusty cores were not observed in other core myopathies and centronuclear myopathies. The other morphological groups in our cohort of patients were: Central Core (CCD: 21%), Core-Rod (C&R:15%) and Type1 predominance "plus" (T1P+:10%). DuCD group was associated to an earlier disease onset, a more severe clinical phenotype and a lowest level of RyR1 expression in muscle, compared to the other groups. Variants located in the bridge solenoid and the pore domains were more frequent in DuCD patients. In conclusion, DuCD is the most frequent histopathological presentation of RYR1-recessive myopathies. Dusty cores represent the unifying morphological lesion among the DuCD pathology spectrum and are the morphological hallmark for the recessive form of disease.
Keywords: Central Core Disease; Centronuclear myopathy; Congenital Myopathy; Dusty Core Disease; RYR1 recessive; Ryanodine receptor.
Conflict of interest statement
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Abath Neto O, Moreno C de AM, Malfatti E, Donkervoort S, Böhm J, Guimarães JB, Foley AR, Mohassel P, Dastgir J, Bharucha-Goebel DX, Monges S, Lubieniecki F, Collins J, Medne L, Santi M, Yum S, Banwell B, Salort-Campana E, Rendu J, Fauré J, Yis U, Eymard B, Cheraud C, Schneider R, Thompson J, Lornage X, Mesrob L, Lechner D, Boland A, Deleuze J-F, Reed UC, Oliveira ASB, Biancalana V, Romero NB, Bönnemann CG, Laporte J, Zanoteli E. Common and variable clinical, histological, and imaging findings of recessive RYR1-related centronuclear myopathy patients. Neuromuscul Disord. 2017;27:975–985. doi: 10.1016/j.nmd.2017.05.016. - DOI - PubMed
-
- Amburgey K, Bailey A, Hwang JH, Tarnopolsky MA, Bonnemann CG, Medne L, Mathews KD, Collins J, Daube JR, Wellman GP, Callaghan B, Clarke NF, Dowling JJ. Genotype-phenotype correlations in recessive RYR1-related myopathies. Orphanet J Rare Dis. 2013;8:117. doi: 10.1186/1750-1172-8-117. - DOI - PMC - PubMed
-
- Bevilacqua JA, Monnier N, Bitoun M, Eymard B, Ferreiro A, Monges S, Lubieniecki F, Taratuto AL, Laquerrière A, Claeys KG, Marty I, Fardeau M, Guicheney P, Lunardi J, Romero NB. Recessive RYR1 mutations cause unusual congenital myopathy with prominent nuclear internalization and large areas of myofibrillar disorganization. Neuropathol Appl Neurobiol. 2011;37:271–284. doi: 10.1111/j.1365-2990.2010.01149.x. - DOI - PubMed
-
- Bharucha-Goebel DX, Santi M, Medne L, Zukosky K, Zukosky K, Dastgir J, Shieh PB, Winder T, Tennekoon G, Finkel RS, Dowling JJ, Monnier N, Bönnemann CG. Severe congenital RYR1-associated myopathy: the expanding clinicopathologic and genetic spectrum. Neurology. 2013;80:1584–1589. doi: 10.1212/WNL.0b013e3182900380. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
