

Vitamin D3 activates the autolysosomal degradation function against Helicobacter pylori through the PDIA3 receptor in gastric epithelial cells
- PMID: 30612517
- PMCID: PMC6526874
- DOI: 10.1080/15548627.2018.1557835
Vitamin D3 activates the autolysosomal degradation function against Helicobacter pylori through the PDIA3 receptor in gastric epithelial cells
Abstract
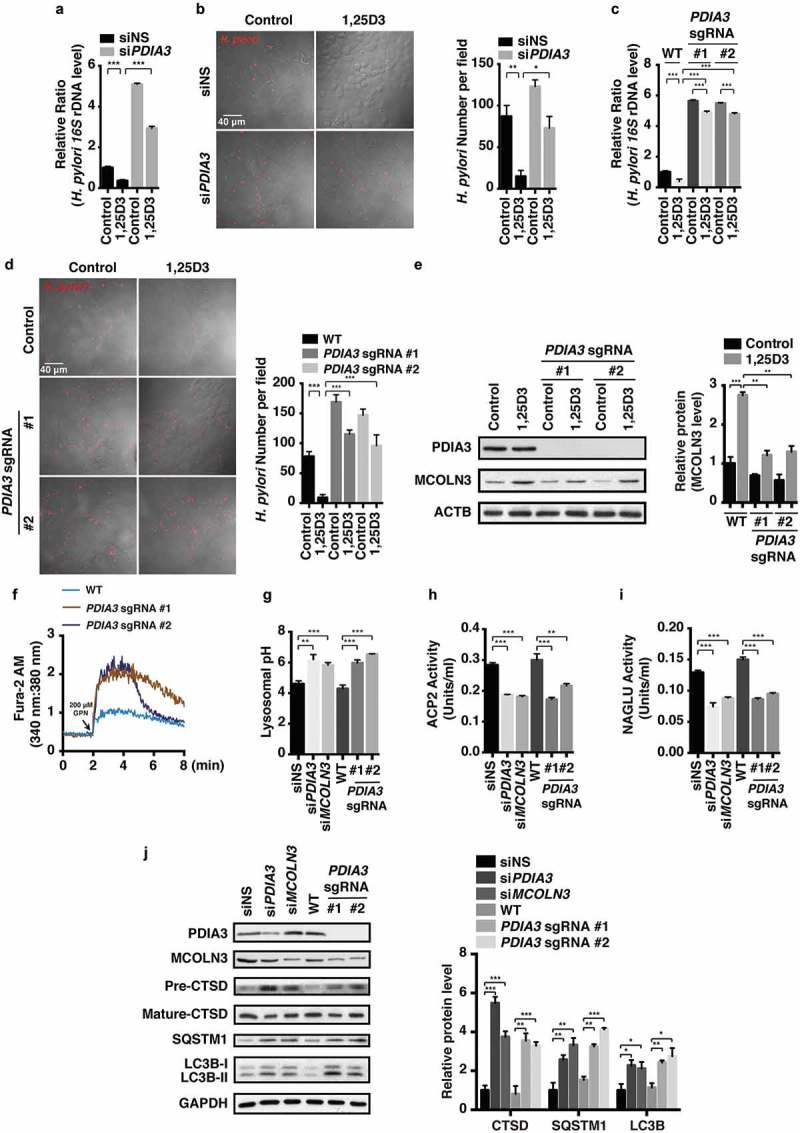
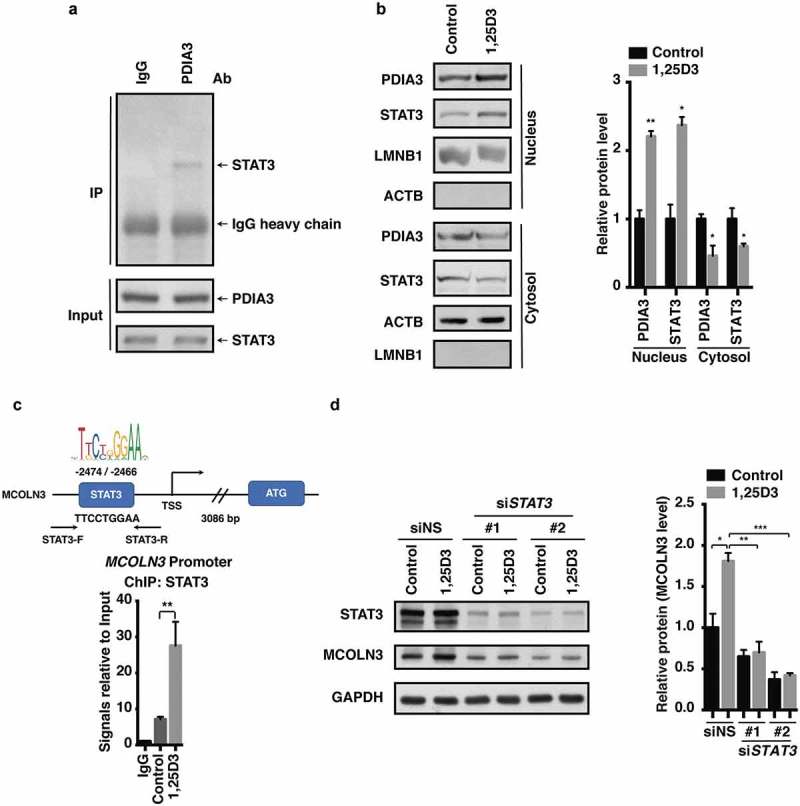
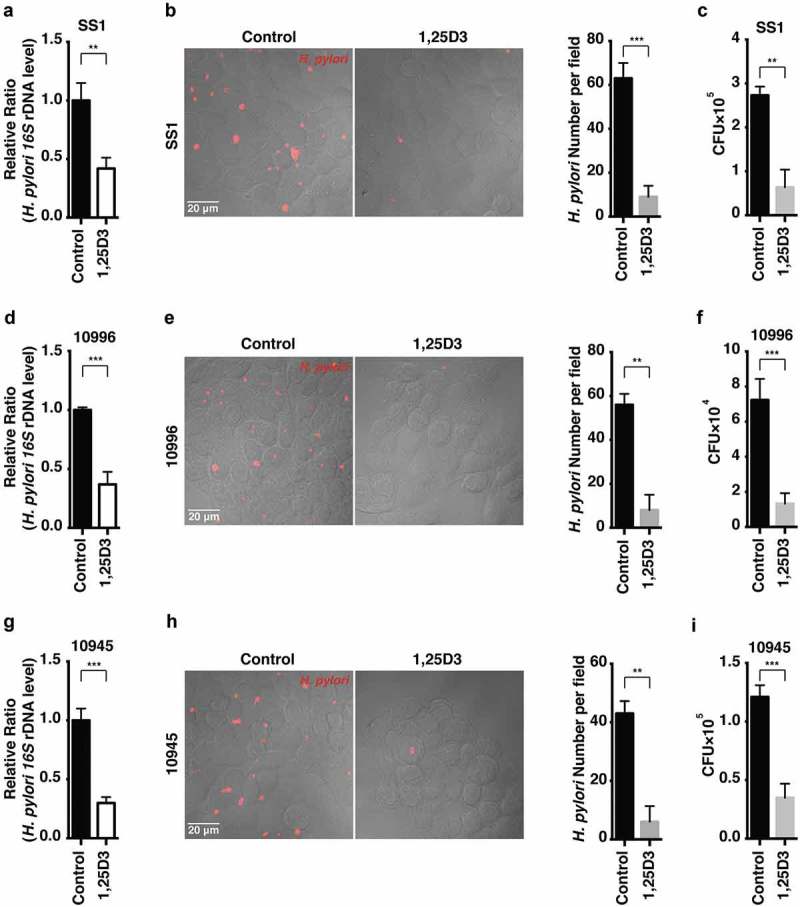
Helicobacter pylori (H. pylori) is a common human pathogenic bacterium. Once infected, it is difficult for the host to clear this organism using the innate immune system. Increased antibiotic resistance further makes it challenging for effective eradication. However, the mechanisms of immune evasion still remain obscure, and novel strategies should be developed to efficiently eliminate H. pylori infection in stomachs. Here we uncovered desirable anti-H. pylori effect of vitamin D3 both in vitro and in vivo, even against antibiotic-resistant strains. We showed that H. pylori can invade into the gastric epithelium where they became sequestered and survived in autophagosomes with impaired lysosomal acidification. Vitamin D3 treatment caused a restored lysosomal degradation function by activating the PDIA3 receptor, thereby promoting the nuclear translocation of PDIA3-STAT3 protein complex and the subsequent upregulation of MCOLN3 channels, resulting in an enhanced Ca2+ release from lysosomes and normalized lysosomal acidification. The recovered lysosomal degradation function drives H. pylori to be eliminated through the autolysosomal pathway. These findings provide a novel pathogenic mechanism on how H. pylori can survive in the gastric epithelium, and a unique pathway for vitamin D3 to reactivate the autolysosomal degradation function, which is critical for the antibacterial action of vitamin D3 both in cells and in animals, and perhaps further in humans. Abbreviations: 1,25D3: 1α, 25-dihydroxyvitamin D3; ATG5: autophagy related 5; Baf A1: bafilomycin A1; BECN1: beclin 1; CagA: cytotoxin-associated gene A; CFU: colony-forming unit; ChIP-PCR: chromatin immunoprecipitation-polymerase chain reaction; Con A: concanamycin A; CQ: chloroquine; CRISPR: clustered regularly interspaced short palindromic repeats; CTSD: cathepsin D; GPN: Gly-Phe-β-naphthylamide; H. pylori: Helicobacter pylori; LAMP1: lysosomal associated membrane protein 1; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MCOLN1: mucolipin 1; MCOLN3: mucolipin 3; MCU: mitochondrial calcium uniporter; MOI: multiplicity of infection; NAGLU: N-acetyl-alpha-glucosaminidase; PDIA3: protein disulfide isomerase family A member 3; PMA: phorbol 12-myristate 13-acetate; PRKC: protein kinase C; SQSTM1: sequestosome 1; STAT3: signal transducer and activator of transcription 3; SS1: Sydney Strain 1; TRP: transient receptor potential; VacA: vacuolating cytotoxin; VD3: vitamin D3; VDR: vitamin D receptor.
Keywords: Autophagy; PDIA3; calcium; lysosome; vitamin D3.
Figures
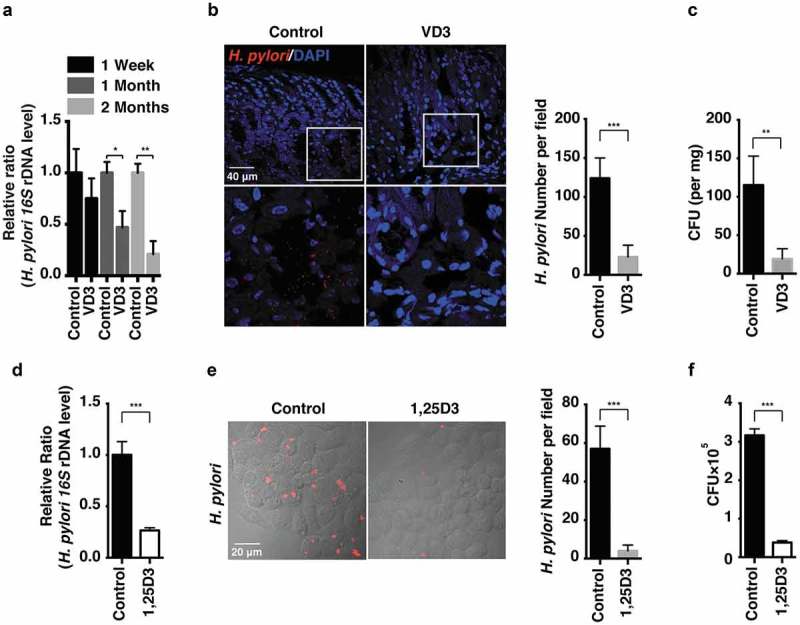
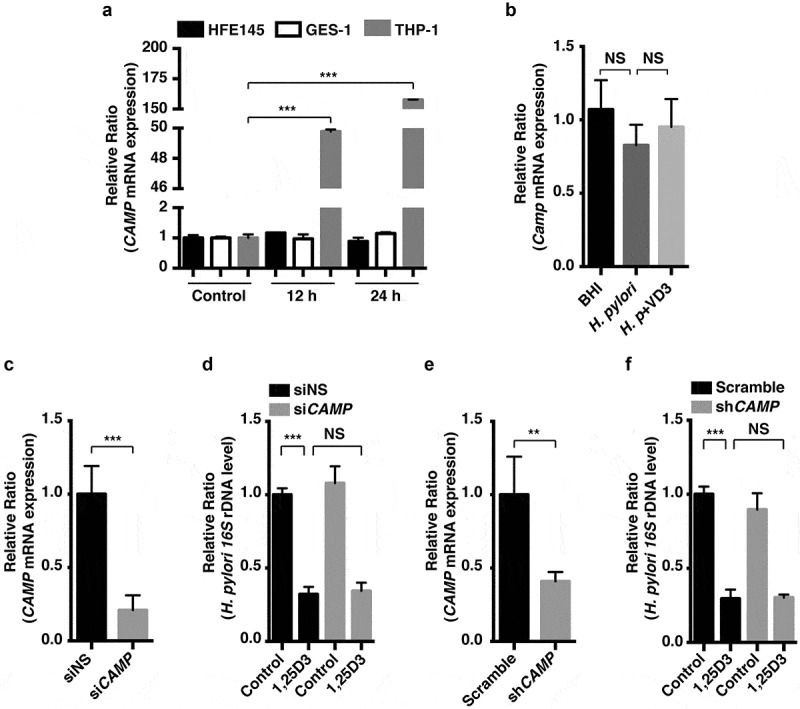
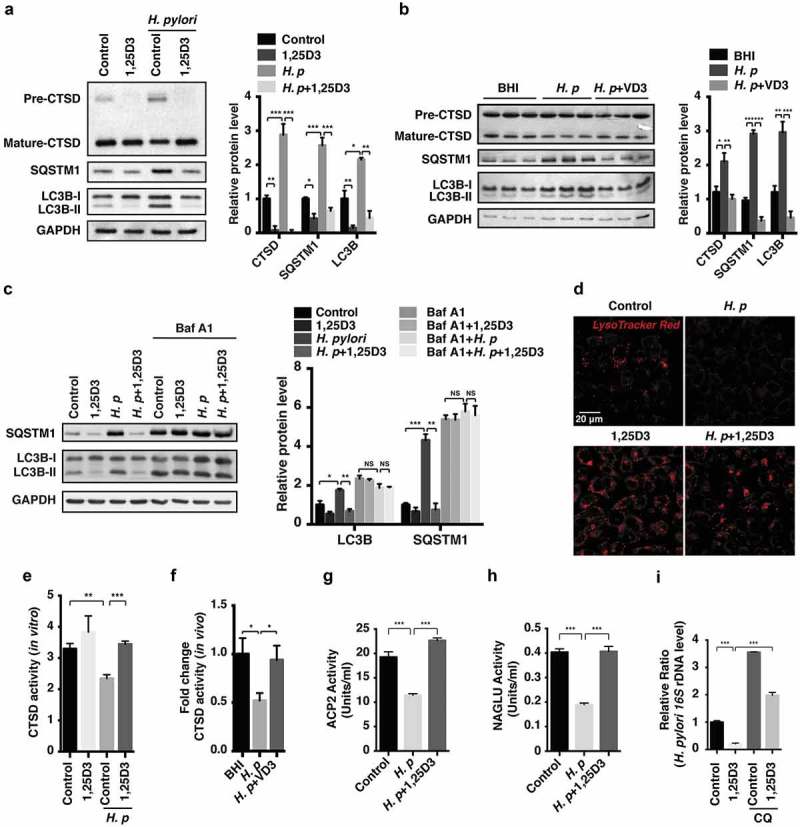
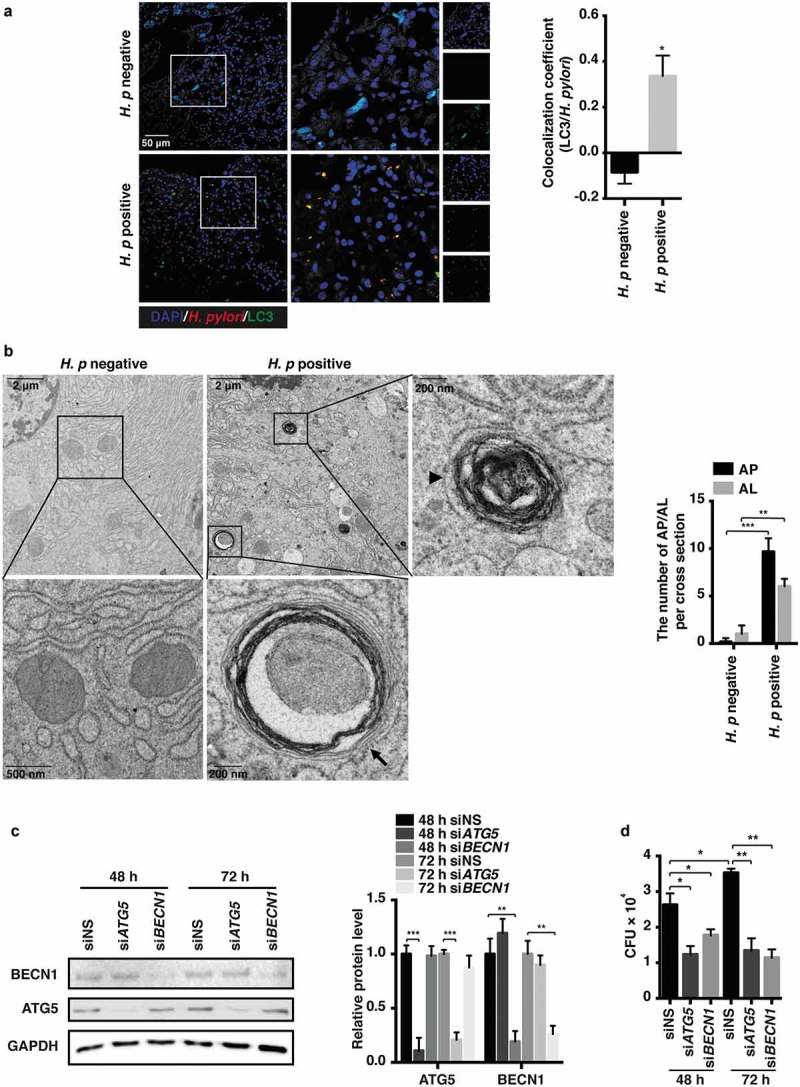


References
-
- Fukase K, Kato M, Kikuchi S, et al. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: an open-label, randomised controlled trial. Lancet. 2008. August 2;372(9636):392–397. PubMed PMID: 18675689. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous