

What Makes a Kinase Promiscuous for Inhibitors?
- PMID: 30612951
- PMCID: PMC6632086
- DOI: 10.1016/j.chembiol.2018.11.005
What Makes a Kinase Promiscuous for Inhibitors?
Abstract
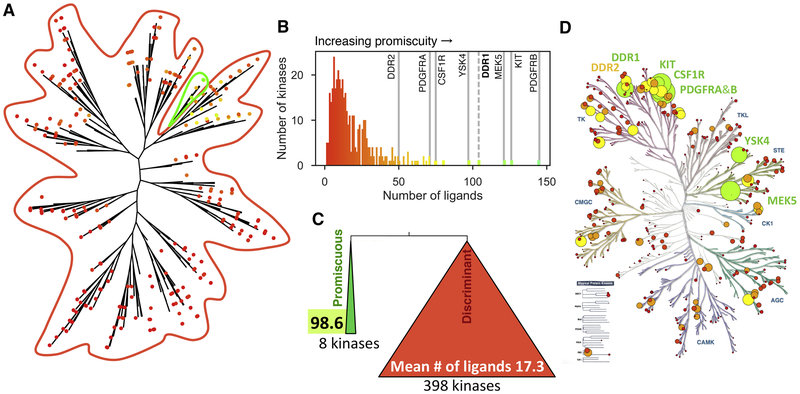
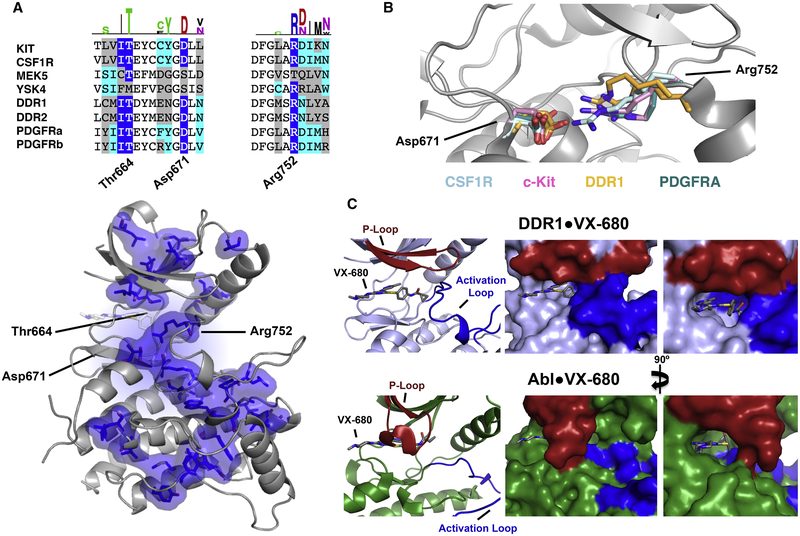
ATP-competitive kinase inhibitors often bind several kinases due to the high conservation of the ATP binding pocket. Through clustering analysis of a large kinome profiling dataset, we found a cluster of eight promiscuous kinases that on average bind more than five times more kinase inhibitors than the other 398 kinases in the dataset. To understand the structural basis of promiscuous inhibitor binding, we determined the co-crystal structure of the receptor tyrosine kinase DDR1 with the type I inhibitors dasatinib and VX-680. Surprisingly, we find that DDR1 binds these type I inhibitors in an inactive conformation typically reserved for type II inhibitors. Our computational and biochemical studies show that DDR1 is unusually stable in this inactive conformation, giving a mechanistic explanation for inhibitor promiscuity. This phenotypic clustering analysis provides a strategy to obtain functional insights not available by sequence comparison alone.
Keywords: ABL; DDR1; DFG; Folding@home; Markov state model; crystallography; drug promiscuity; kinase inhibition; molecular dynamics; selectivity.
Copyright © 2018 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests
J.D.C. is a member of the S.A.B. of Schrödinger LLC.
Figures


References
-
- Adrián FJ, Ding Q, Sim T, Velentza A, Sloan C, Liu Y, Zhang G, Hur W, Ding S, Manley P, et al. (2006). Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nature Chemical Biology 2, 95–102. - PubMed
-
- Azam M, Latek RR, and Daley GQ (2003). Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell 112, 831–843. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
