

Assessment of circulating tumor DNA in pediatric solid tumors: The promise of liquid biopsies
- PMID: 30614191
- PMCID: PMC6550461
- DOI: 10.1002/pbc.27595
Assessment of circulating tumor DNA in pediatric solid tumors: The promise of liquid biopsies
Abstract
Circulating tumor DNA can be detected in the blood and body fluids of patients using ultrasensitive technologies, which have the potential to improve cancer diagnosis, risk stratification, noninvasive tumor profiling, and tracking of treatment response and disease recurrence. As we begin to apply "liquid biopsy" strategies in children with cancer, it is important to tailor our efforts to the unique genomic features of these tumors and address the technical and logistical challenges of integrating biomarker testing. This article reviews the literature demonstrating the feasibility of applying liquid biopsy to pediatric solid malignancies and suggests new directions for future studies.
Keywords: circulating tumor DNA; digital PCR; liquid biopsy; next-generation sequencing; pediatric cancer.
© 2019 Wiley Periodicals, Inc.
Conflict of interest statement
Conflict of Interest statement: The authors declare that there is no conflict of interest regarding the publication of this article.
Figures
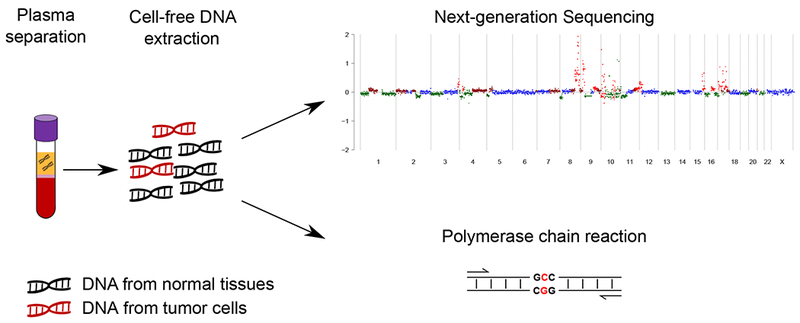
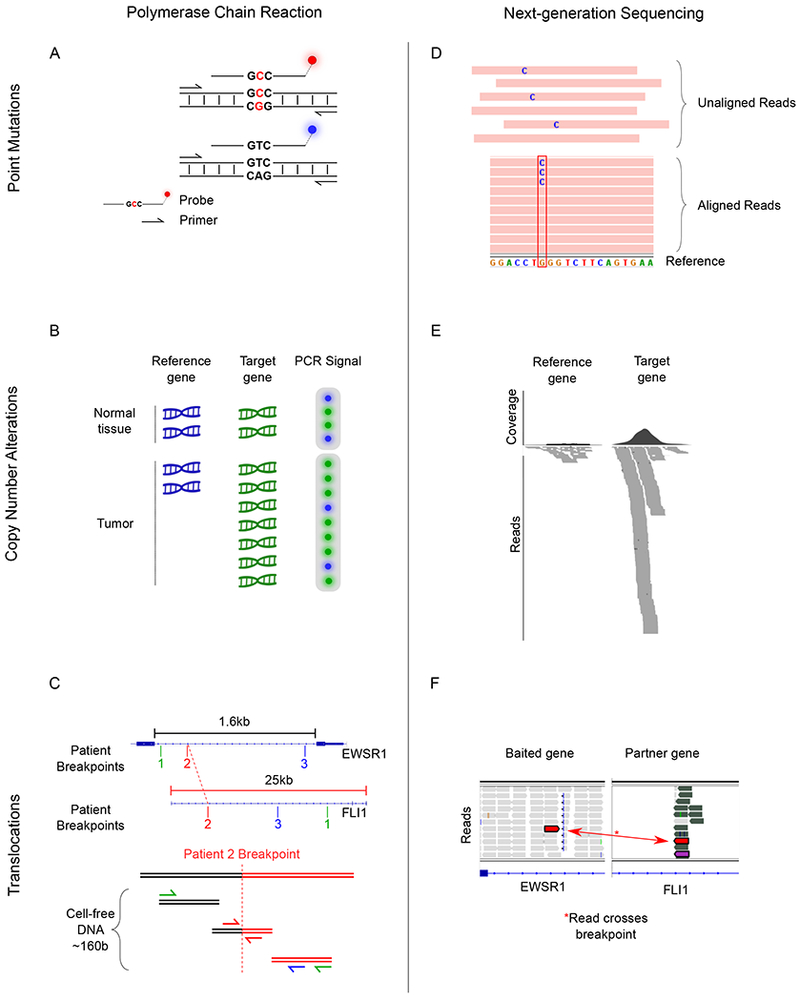
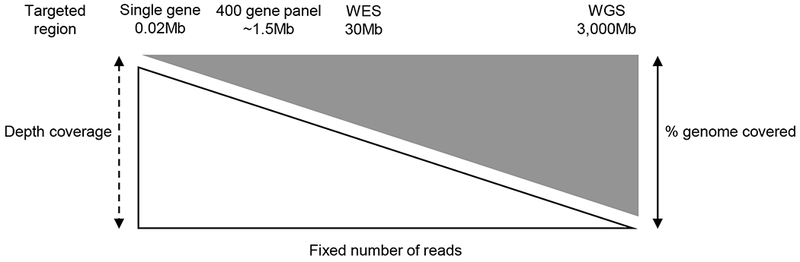
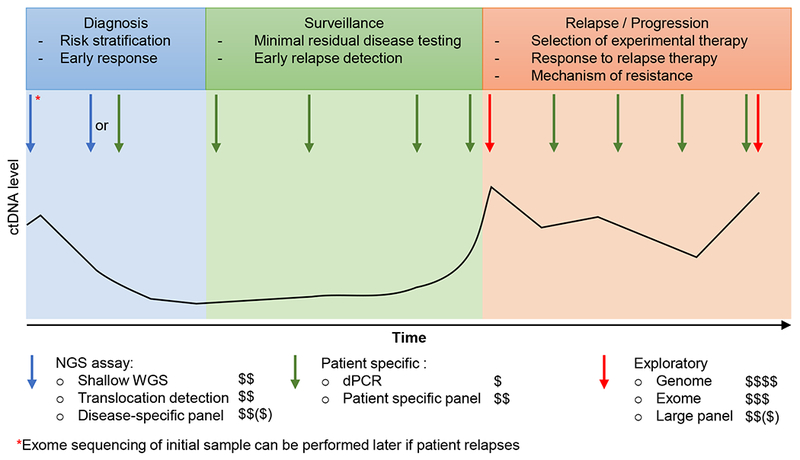
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
