

Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis
- PMID: 30616300
- PMCID: PMC6353056
- DOI: 10.1002/14651858.CD009841.pub3
Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis
Abstract
Background: Cystic fibrosis (CF) is the commonest inherited life-shortening illness in white populations, caused by a mutation in the gene that codes for the cystic fibrosis transmembrane regulator protein (CFTR), which functions as a salt transporter. This mutation mainly affects the airways where excess salt absorption dehydrates the airway lining leading to impaired mucociliary clearance. Consequently, thick, sticky mucus accumulates making the airway prone to chronic infection and progressive inflammation; respiratory failure often ensues. Other complications include malnutrition, diabetes and subfertility.Increased understanding of the condition has allowed pharmaceutical companies to design mutation-specific therapies targeting the underlying molecular defect. CFTR potentiators target mutation classes III and IV and aim to normalise airway surface liquid and mucociliary clearance, which in turn impacts on the chronic infection and inflammation. This is an update of a previously published review.
Objectives: To evaluate the effects of CFTR potentiators on clinically important outcomes in children and adults with CF.
Search methods: We searched the Cochrane Cystic Fibrosis Trials Register, compiled from electronic database searches and handsearching of journals and conference abstract books. We also searched the reference lists of relevant articles, reviews and online clinical trial registries. Last search: 21 November 2018.
Selection criteria: Randomised controlled trials (RCTs) of parallel design comparing CFTR potentiators to placebo in people with CF. A separate review examines trials combining CFTR potentiators with other mutation-specific therapies.
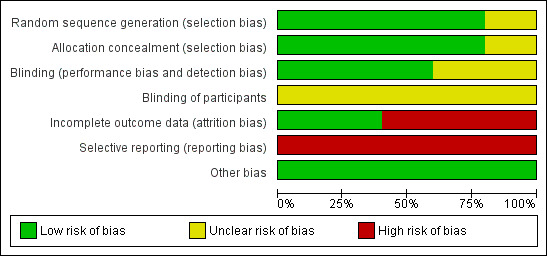
Data collection and analysis: The authors independently extracted data, assessed the risk of bias in included trials and used GRADE to assess evidence quality. Trial authors were contacted for additional data.
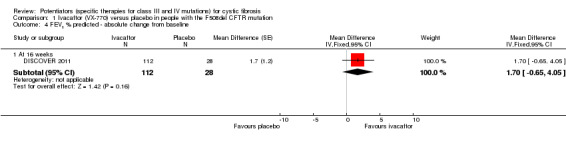
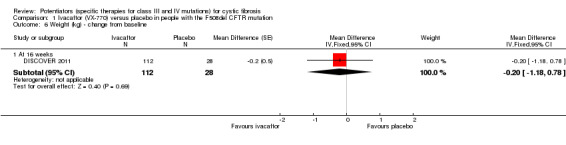
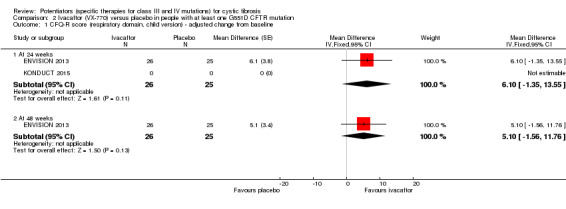
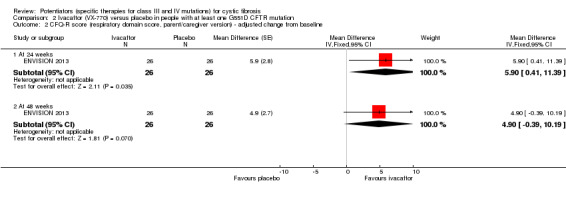
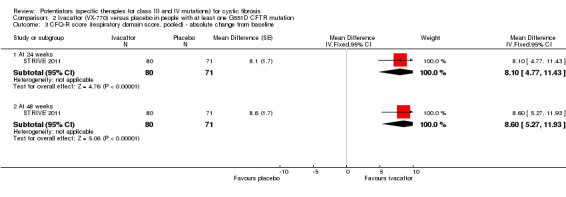
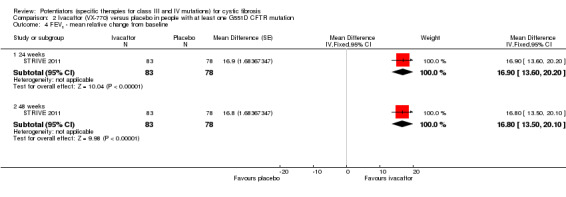
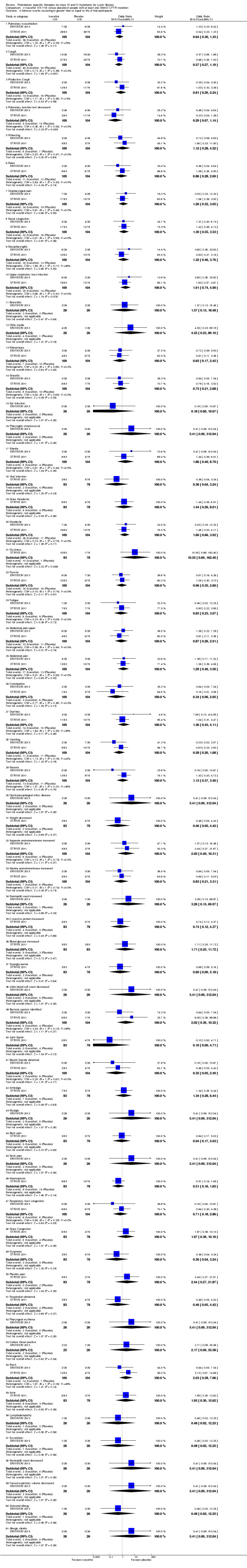
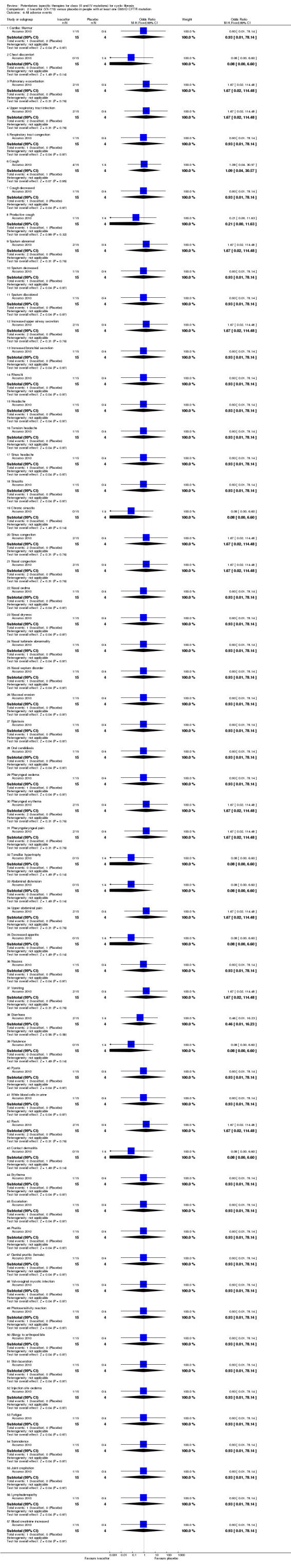
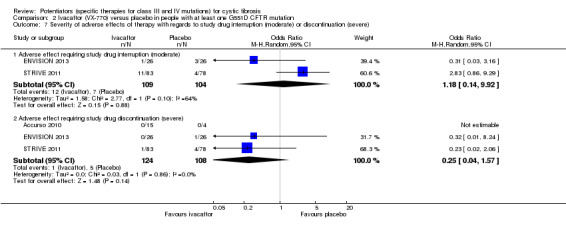
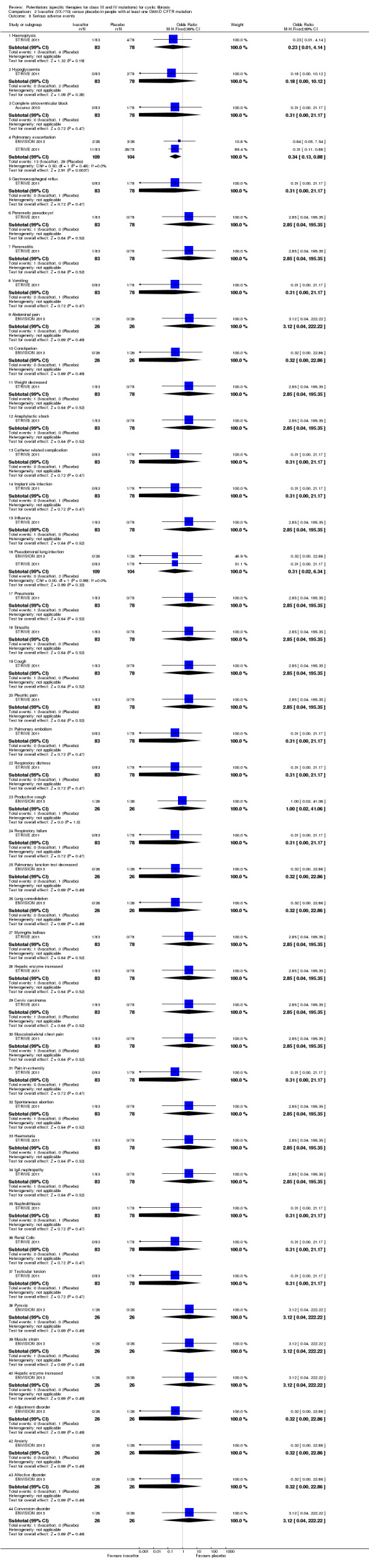
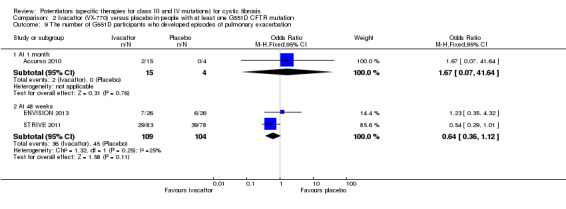
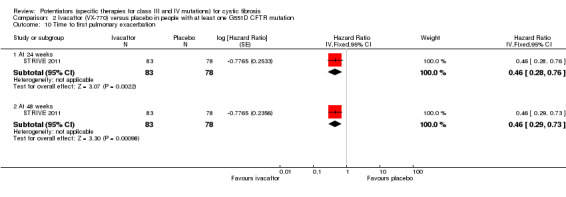
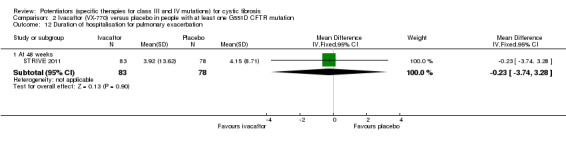
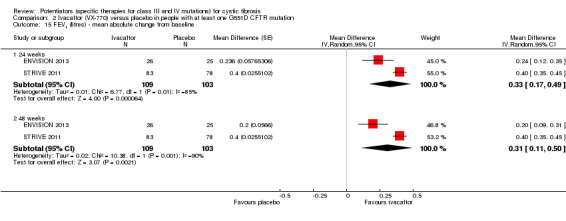
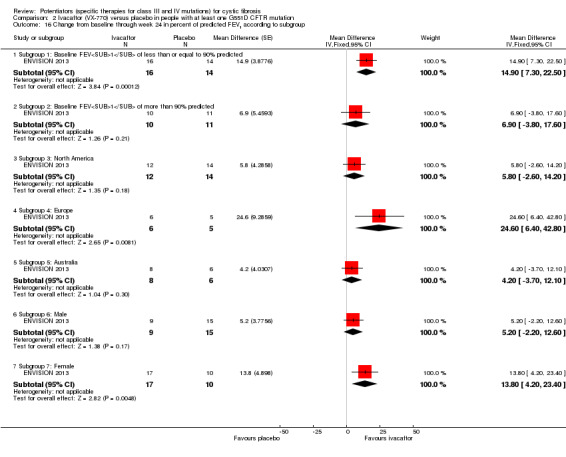
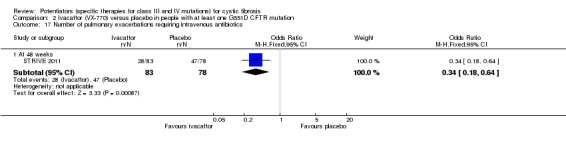
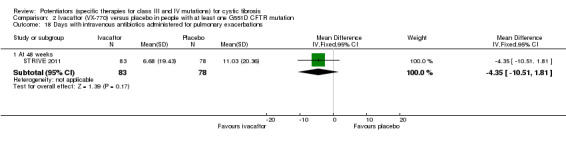
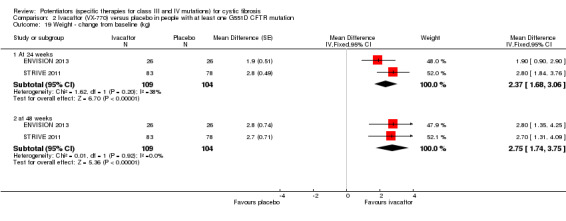
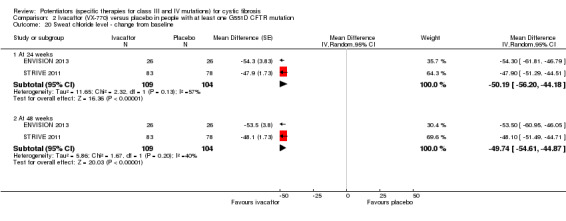
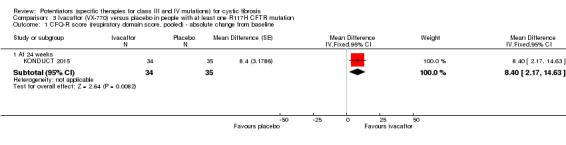
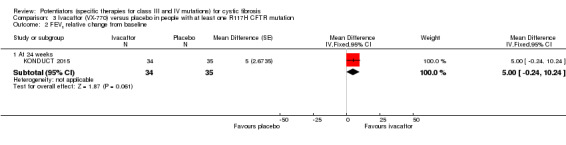
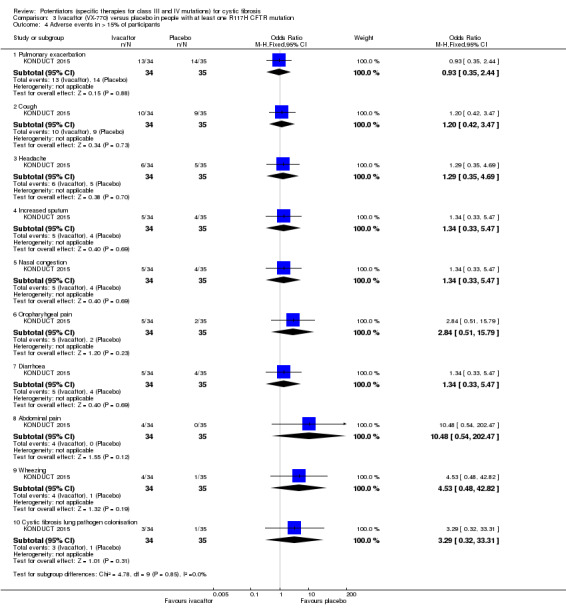
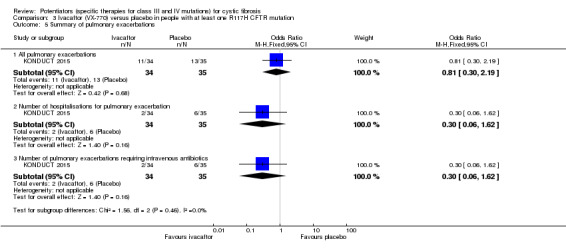
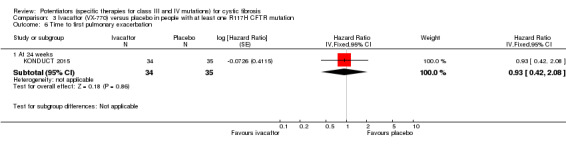
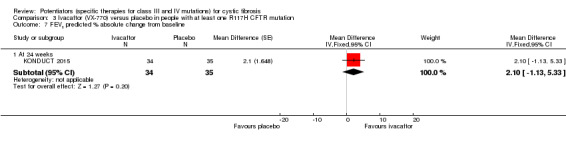
Main results: We included five RCTs (447 participants with different mutations) lasting from 28 days to 48 weeks, all assessing the CFTR potentiator ivacaftor. The quality of the evidence was moderate to low, mainly due to risk of bias (incomplete outcome data and selective reporting) and imprecision of results, particularly where few individuals experienced adverse events. Trial design was generally well-documented. All trials were industry-sponsored and supported by other non-pharmaceutical funding bodies.F508del (class II) (140 participants)One 16-week trial reported no deaths, or changes in quality of life (QoL) or lung function (either relative or absolute change in forced expiratory volume in one second (FEV1) (moderate-quality evidence). Pulmonary exacerbations and cough were the most reported adverse events in ivacaftor and placebo groups, but there was no difference between groups (low-quality evidence); there was also no difference between groups in participants interrupting or discontinuing treatment (low-quality evidence). Number of days until the first exacerbation was not reported, but there was no difference between groups in how many participants developed pulmonary exacerbations. There was also no difference in weight. Sweat chloride concentration decreased, mean difference (MD) -2.90 mmol/L (95% confidence interval (CI) -5.60 to -0.20).G551D (class III) (238 participants)The 28-day phase 2 trial (19 participants) and two 48-week phase 3 trials (adult trial (167 adults), paediatric trial (52 children)) reported no deaths. QoL scores (respiratory domain) were higher with ivacaftor in the adult trial at 24 weeks, MD 8.10 (95% CI 4.77 to 11.43) and 48 weeks, MD 8.60 (95% CI 5.27 to 11.93 (moderate-quality evidence). The adult trial reported a higher relative change in FEV1 with ivacaftor at 24 weeks, MD 16.90% (95% CI 13.60 to 20.20) and 48 weeks, MD 16.80% (95% CI 13.50 to 20.10); the paediatric trial reported this at 24 weeks, MD 17.4% (P < 0.0001)) (moderate-quality evidence). These trials demonstrated absolute improvements in FEV1 (% predicted) at 24 weeks, MD 10.80% (95% CI 8.91 to 12.69) and 48 weeks, MD 10.44% (95% CI 8.56 to 12.32). The phase 3 trials reported increased cough, odds ratio (OR) 0.57 (95% CI 0.33 to 1.00) and episodes of decreased pulmonary function, OR 0.29 (95% CI 0.10 to 0.82) in the placebo group; ivacaftor led to increased dizziness in adults, OR 10.55 (95% CI 1.32 to 84.47). There was no difference between groups in participants interrupting or discontinuing treatment (low-quality evidence). Fewer participants taking ivacaftor developed serious pulmonary exacerbations; adults taking ivacaftor developed fewer exacerbations (serious or not), OR 0.54 (95% CI 0.29 to 1.01). A higher proportion of participants were exacerbation-free at 24 weeks with ivacaftor (moderate-quality evidence). Ivacaftor led to a greater absolute change from baseline in FEV1 (% predicted) at 24 weeks, MD 10.80% (95% CI 8.91 to 12.69) and 48 weeks, MD 10.44% (95% CI 8.56 to 12.32); weight also increased at 24 weeks, MD 2.37 kg (95% CI 1.68 to 3.06) and 48 weeks, MD 2.75 kg (95% CI 1.74 to 3.75). Sweat chloride concentration decreased at 24 weeks, MD -48.98 mmol/L (95% CI -52.07 to -45.89) and 48 weeks, MD -49.03 mmol/L (95% CI -52.11 to -45.94).R117H (class IV) (69 participants)One 24-week trial reported no deaths. QoL scores (respiratory domain) were higher with ivacaftor at 24 weeks, MD 8.40 (95% CI 2.17 to 14.63), but no relative changes in lung function were reported (moderate-quality evidence). Pulmonary exacerbations and cough were the most reported adverse events in both groups, but there was no difference between groups; there was no difference between groups in participants interrupting or discontinuing treatment (low-quality evidence). Number of days until the first exacerbation was not reported, but there was no difference between groups in how many participants developed pulmonary exacerbations. No changes in absolute change in FEV1 or weight were reported. Sweat chloride concentration decreased, MD -24.00 mmol/L (CI 95% -24.69 to -23.31).
Authors' conclusions: There is no evidence supporting the use of ivacaftor in people with the F508del mutation. Both G551D phase 3 trials demonstrated a clinically relevant impact of ivacaftor on outcomes at 24 and 48 weeks in adults and children (over six years of age) with CF. The R117H trial demonstrated an improvement in the respiratory QoL score, but no improvement in respiratory function.As new mutation-specific therapies emerge, it is important that trials examine outcomes relevant to people with CF and their families and that adverse events are reported robustly and consistently. Post-market surveillance is essential and ongoing health economic evaluations are required.
Conflict of interest statement
Mica Skilton declares no potential conflict of interest.
Ian Sinha declares no potential conflict of interest.
Sanjay Patel declares no potential conflict of interest.
Kevin Southern declares no potential conflict of interest.
Figures
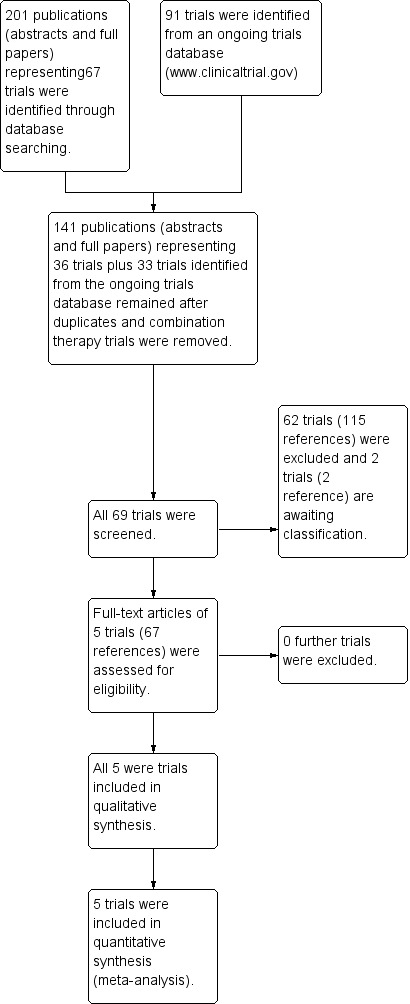












Update of
-
Potentiators (specific therapies for class III and IV mutations) for cystic fibrosis.Cochrane Database Syst Rev. 2015 Mar 26;(3):CD009841. doi: 10.1002/14651858.CD009841.pub2. Cochrane Database Syst Rev. 2015. Update in: Cochrane Database Syst Rev. 2019 Jan 07;1:CD009841. doi: 10.1002/14651858.CD009841.pub3. PMID: 25811419 Updated.
References
References to studies included in this review
Accurso 2010 {published data only}
-
- Accurso F, Rowe SM, Durie PR, Konstan MW, Dunitz J, Hornick D, et al. Improvement in sweat chloride concentration by the CFTR potentiator VX‐770 in subjects with cystic fibrosis and the G551D‐CFTR mutation. Pediatric Pulmonology 2009;44(S32):296. [Abstract no.: 240; CFGD Register: BD165c; MEDLINE: ]
-
- Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Online supplementary material (disclosure forms) to "Effect of VX‐770 in persons with cystic fibrosis and the G551D‐CFTR mutation" [online]. New England Journal of Medicine 2010;363(21):1991‐2003 online. [CFGD Register: BD165k; MEDLINE: ] - PMC - PubMed
-
- Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, et al. Online supplementary material (protocol) to "Effect of VX‐770 in persons with cystic fibrosis and the G551D‐CFTR mutation" [online]. New England Journal of Medicine 2010;363(21):1‐121 online. [CFGD Register: BD165l; MEDLINE: ] - PMC - PubMed
DISCOVER 2011 {published data only}
-
- Flume PA, Borowitz D, Liou T, Li H, Yen K, Ordonez C, et al. VX‐770 in subjects with CF and homozygous for the F508del‐CFTR mutation. Pediatric Pulmonology 2011;46 Suppl 34:284. [Abstract no.: 206; CFGD Register: BD168b; MEDLINE: ]
ENVISION 2013 {published data only}
-
- Aherns R, Rodriguez S, Yen K, Davies JC. VX‐770 in subjects 6 to 11 years with cystic fibrosis and the G551D‐CFTR mutation. Pediatric Pulmonology 2011;46(S34):283. [Abstract no.: 203; CFGD Register: BD171a; MEDLINE: ]
-
- Borowitz D, Lubarsky B, Wilschanski M, Munck A, Gelfond D, Bodewes F, et al. Nutritional status improved in cystic fibrosis patients with the G551D mutation. Digestive Diseases and Sciences 2016;61(1):198‐207. [CFGD Register: BD170u // BD171p] - PubMed
-
- Borowitz D, Ramsey B, Dong Q, Yen K, Elborn JS. Measures of nutritional status in two Phase 3 trials of ivacaftor in subjects with cystic fibrosis who have the G551D‐CFTR mutation. Journal of Cystic Fibrosis 2012;11 Suppl 1:S13. [Abstract no.: WS6.3; CFGD Register: BD171c//BD170d; ]
-
- Borowitz D, Ramsey B, Rodriguez S, Yen K, Elborn JS. Nutritional status measures among persons with CF carrying the G551D‐CFTR mutation who received ivacaftor or placebo in phase 3 clinical trials. Pediatric Pulmonology 2012;47(S35):298. [Abstract no.: 214; CFGD Register: BD171f//BD170i; ]
-
- Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. American Journal of Respiratory and Critical Care Medicine 2013;187(11):1219‐25. [CFGD Register: BD171g; ] - PMC - PubMed
KONDUCT 2015 {published data only}
-
- Moss R. Study of ivacaftor in subjects with cystic fibrosis who have the R117H‐CFTR mutation (KONDUCT). Www.clinicaltrials.gov (www.clinicaltrials.gov) (accessed 22 Oct 2014) 2014. [CENTRAL: 1012721; CFGD Register: BD204b; CRS: 5500131000000199; NCT01614457]
-
- Moss R, Flume PA, Elborn J, Cooke J, Rowe SM, McColley SA, et al. Effects of ivacaftor in CF patients with R117H‐CFTR. Pediatric Pulmonology 2014;49 Suppl 38:221. [Abstract no.: 17; CENTRAL: 1012722; CFGD Register: BD204c; CRS: 5500131000000201]
-
- Moss RB, Flume PA, Elborn JS, Cooke J, Rowe SM, McColley SA, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis who have an Arg117His‐CFTR mutation: a double‐blind, randomised controlled trial. The Lancet. Respiratory Medicine 2015;3(7):524‐33. [CFGD Register: BD204d] - PMC - PubMed
-
- Moss RB, Flume PA, Elborn JS, Cooke J, Rowe SM, McColley SA, et al. Ivacaftor treatment in patients with cystic fibrosis who have an R117H‐CFTR mutation, the KONDUCT study. Journal of Cystic Fibrosis 2014;13 Suppl 2:S44. [Abstract no.: WS23.6; CENTRAL: 996577; CFGD Register: BD204a; CRS: 5500129000000012]
STRIVE 2011 {published data only}
-
- Borowitz D, Lubarsky B, Wilschanski M, Munck A, Gelfond D, Bodewes F, et al. Nutritional status improved in cystic fibrosis patients with the G551D mutation. Digestive Diseases and Sciences 2016;61(1):198‐207. [CFGD Register: BD170u // BD171p] - PubMed
-
- Borowitz D, Ramsey B, Dong Q, Yen K, Elborn JS. Measures of nutritional status in two Phase 3 trials of ivacaftor in subjects with cystic fibrosis who have the G551D‐CFTR mutation. Journal of Cystic Fibrosis 2012;11 Suppl 1:S13. [Abstract no.: WS6.3; CFGD Register: BD170d//BD171c; ]
-
- Borowitz D, Ramsey B, Rodriguez S, Yen K, Elborn JS. Nutritional status measures among persons with CF carrying the G551D‐CFTR mutation who received ivacaftor or placebo in phase 3 clinical trials. Pediatric Pulmonology 2012;47 Suppl 35:298. [Abstract no.: 214; CFGD Register: BD170i//BD171f; ]
-
- Elborn JS, Rodriguez S, Lubarsky B, Gilmartin G, Bell S. Effect of ivacaftor in patients with cystic fibrosis and the G551D‐CFTR mutation who have baseline FEV >90% of predicted. Pediatric Pulmonology 2013;48 Suppl 36:298. [Abstract no.: 257; CENTRAL: 921689; CFGD Register: BD170m // BD171k // BD188e ; CRS: 5500125000000401]
-
- Elborn S, Plant B, Konstan M, Aherns R, Rodriguez S, Munck A, et al. Lung function, weight, and sweat chloride responses in patients with cystic fibrosis and the G551D‐CFTR mutation treated with ivacaftor: A secondary analysis. European Respiratory Journal 2013;42:1073s. [Abstract no.: 5059; CENTRAL: 1099910; CFGD Register: BD170o // BD171n ; CRS: 5500050000000278; EMBASE: 71843034]
References to studies excluded from this review
Accurso 2013 {published data only}
-
- Accurso FJ, Ratjen F, Altes T, Lubarsky B, Dong Q, Kang L, et al. Effect of withdrawal of ivacaftor therapy on CFTR channel activity and lung function in patients with cystic fibrosis. Journal of Cystic Fibrosis 2013;12 Suppl 1:S62. [Abstract no.: 56; CFGD Register: BD189//BD188c; ]
ALBATROSS 2017 {published data only}
-
- Bell S, Boeck K, Drevinek P, Plant B, Barry P, Elborn S, et al. GLPG2222 in subjects with cystic fibrosis and the F508del/Class III mutation on stable treatment with ivacaftor: results from a phase II study (ALBATROSS). Journal of cystic fibrosis 2018;17(Suppl 3):S2. [CFGD Register: BD247a; clinicaltrials.gov: NCT03045523]
-
- Bell SC, Boeck K, Drevinek P, Plant BJ, Elborn J, Kock H, et al. Results from a phase ii study ‐ albatross ‐ evaluation of glpg2222 in subjects with cf and the f508del/class iii mutation on stable treatment with ivacaftor. Pediatric Pulmonology 2018;53(S2):249. [CFGD Register: BD247b; clinicaltrials.gov: NCT03045523]
Altes 2011 {published data only}
-
- Altes T, Johnson M, Higgins M, Fidler M, Botfield M, Mugler III JP, et al. The effect of ivacaftor treatment on lung ventilation defects, as measured by hyperpolarized helium‐3 MRI, on patients with cystic fibrosis and a G551D‐CFTR mutation. Journal of Cystic Fibrosis 2014;13 Suppl 2:S6. [Abstract no.: WS3.2; CENTRAL: 1000056; CFGD Register: BD172d; CRS: 5500131000000009]
-
- Altes T, Johnson M, Higgins M, Fidler M, Botfield M, Tustison N, Leiva‐Salinas C. Use of hyperpolarized helium‐3 MRI to assess response to ivacaftor treatment in patients with cystic fibrosis. Journal of Cystic Fibrosis 2017;16:267‐274. - PubMed
-
- Altes T, Johnson MA, Miller GW, Mugler JP, Flors L, Mata J, et al. Hyperpolarized Gas MRI of ivacaftor therapy in subjects with cystic fibrosis who have the G551D‐CFTR mutation. Journal of Cystic Fibrosis 2012;11 Suppl 1:S67. [Abstract no.: 46; CFGD Register: BD172b; ]
-
- Altes T, Johnson MA, Miller GW, Mugler JP, Flors L, Mata J, et al. Hyperpolarized gas MRI of ivacaftor therapy in persons with cystic fibrosis and the G551D‐CFTR mutation. Pediatric Pulmonology 2012;47 Suppl 35:291. [Abstract no.: 196; CFGD Register: BD172c; ]
-
- Altes T, Johnson MA, Miller GW, Mugler JP, Flors L, Mata J, et al. Hyperpolarized helium‐3 magnetic resonance imaging of CFTR potentiator therapy in subjects with cystic fibrosis and the G551D mutation. Pediatric Pulmonology 2011;46(S34):284. [Abstract no.: 205; CFGD Register: BD172a; MEDLINE: ]
ARRIVAL 2018 {published data only}
-
- Rosenfeld M, Wainwright CE, Higgins M, Wang LT, McKee C, Campbell D, et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single‐arm study. The Lancet. Respiratory Medicine 2018;6(7):545‐53. [PUBMED: 29886024] - PMC - PubMed
Berkers 2017 {published data only}
-
- Berkers G, Mourik P, Dekkers JF, Kruisselbrink E, Vonk AM, Heida‐Michel S, et al. Correlation between individual clinical responses and forskolin‐induced swelling of paired intestinal organoids upon CFTR modulator treatment. Pediatric Pulmonology 2017;52 Suppl 47:295. [CFGD Register: BD242]
Chadwick 1998 {published data only}
-
- Chadwick S, Browning JE, Stern M, Cheng SH, Gruenert DC, Geddes DM, et al. Nasal application of glycerol in DF508 cystic fibrosis patients. Pediatric Pulmonology 1998;26 Suppl 17:278. [CFGD Register: BD147]
Clancy 2012 {published data only}
-
- Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, et al. Results of a phase IIa study of VX‐809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del‐CFTR mutation. Thorax 2012;67(1):12‐8. [CFGD Register: BD166c; MEDLINE: ] - PMC - PubMed
-
- Clancy JP, Rowe SM, Accurso FJ, Ballmann M, Boyle MP, DeBoeck C, et al. A phase II, randomized, placebo‐controlled, clinical trial of four doses of VX‐809 in CF patients homozygous for the F508del CFTR mutation. Pediatric Pulmonology 2010;45 Suppl 33:298. [Abstract no.: 224; CFGD Register: BD166b; MEDLINE: ]
-
- Clancy JP, Rowe SM, Liu B, Hathorne H, Dong Q, Wisseh S, et al. Variability of nasal potential difference measurements in clinical testing of CFTR modulators [abstract]. Pediatric Pulmonology 2011;46 Suppl 34:283, Abstract no: 202. [CFGD Register: BD165n, BD166d; MEDLINE: ]
-
- Clancy JP, Spencer‐Green G, for the VX‐809‐101 Study Group. Clinical evaluation of VX‐809, a novel investigational oral F508del‐CFTR corrector, in subjects with cystic fibrosis homozygous for the F508del‐CFTR mutation. Journal of Cystic Fibrosis 2010;9 Suppl 1:S20. [Abstract no.: 73; CFGD Register: BD166a; MEDLINE: ]
Davies 2012 {published data only}
-
- Accurso FJ, Ratjen F, Altes T, Lubarsky B, Dong Q, Kang L, et al. Effect of withdrawal of ivacaftor therapy on CFTR channel activity and lung function in patients with cystic fibrosis. Journal of Cystic Fibrosis 2013;12 Suppl 1:S62. [Abstract no.: 56; CFGD Register: BD189//BD190b//BD188c; ]
-
- Davies J, Sheridan H, Bell N, Cunningham S, Davis SD, Elborn JS, et al. Assessment of clinical response to ivacaftor with lung clearance index in cystic fibrosis patients with a G551D‐CFTR mutation and preserved spirometry: a randomised controlled trial. The Lancet. Respiratory Medicine 2013;1(8):630‐8. [CFGD Register: BD188f] - PubMed
-
- Davies JC, Sheridan H, Lee P, Song T, Stone A, Ratjen F. Lung clearance index to evaluate the effect of ivacaftor on lung function in subjects with CF who have the G551D‐CFTR mutation and mild lung disease. Pediatric Pulmonology 2012;47 Suppl 35:311. [Abstract no.: 249; CFGD Register: BD188b; ]
-
- Davies JC, Sheridan H, Lee PS, Song T, Stone A, Ratjen F, et al. Effect of ivacaftor on lung function in subjects with CF who have the G551D‐CFTR mutation and mild lung disease: a comparison of lung clearance index (LCI) vs. spirometry. Journal of Cystic Fibrosis 2012;11 Suppl 1:S15. [Abstract no.: WS7.6; CFGD Register: BD188a; ]
-
- Elborn JS, Rodriguez S, Lubarsky B, Gilmartin G, Bell S. Effect of ivacaftor in patients with cystic fibrosis and the G551D‐CFTR mutation who have baseline FEV >90% of predicted. Pediatric Pulmonology 2013;48 Suppl 36:298. [Abstract no.: 257; CENTRAL: 921689; CFGD Register: BD188e // BD170m // BD171k; CRS: 5500125000000401]
Davies 2016 {published data only}
-
- Davies JC, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2‐5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open‐label, single‐arm study. The Lancet. Respiratory medicine 2016;4(2):107‐15. [PUBMED: 26803277] - PMC - PubMed
Edgeworth 2017 {published data only}
-
- Button BM, Edgeworth D, Finlayson F, Fantidis M, Wilson L, Talbot A, et al. Effect of ivacaftor on wellness, quality of life and cognitive function in adults with cystic fibrosis and G551D mutation. Journal of Cystic Fibrosis 2015;14 Suppl 1:S18. [Abstract no.: WS09.5; CENTRAL: 1081475; CFGD Register: BD222a; CRS: 5500135000000012]
-
- Button BM, Edgeworth D, Wilson LM, Sayer J, Tierney A, Finlayson F, et al. Ivacaftor improves wellness, quality of life and cognitive function in G551D cystic fibrosis. Pediatric Pulmonology 2015;50 Suppl 41:437. [Abstract no.: 643; CENTRAL: 1092211; CFGD Register: BD222e; CRS: 5500135000001400]
-
- Edgeworth D, Keating D, Ellis M, Button B, Williams E, Clark D, et al. Improvement in exercise duration, lung function and well‐being in G551D‐cystic fibrosis patients: a double‐blind, placebo‐controlled, randomized, cross‐over study with ivacaftor treatment. Clinical Science 2017;131(15):2037‐45. [CFGD Register: BD222f] - PubMed
-
- Edgeworth D, Keating D, Williams E, Clark D, Button B, Tierney A, et al. Exercise improvements in ivacaftor treated G551D cystic fibrosis patients are not solely related to FEV1 and sweat changes. European Respiratory Journal 2015;46 Suppl:PA2047. [CFGD Register: BD222j; DOI: 10.1183/13993003.congress2015.PA2047] - DOI
-
- Edgeworth D, Keating D, Williams E, Clark D, Button BM, Tierney AC, et al. Ivacaftor improves exercise capacity in patients with G551D CF gene mutations. Journal of Cystic Fibrosis : Official Journal of the European Cystic Fibrosis Society 2015;14 Suppl 1:S27. [Abstract no.: WS14.1; CENTRAL: 1081478; CFGD Register: BD222b; CRS: 5500135000000015]
EudraCT Number: 2014‐000817‐30 {published data only}
-
- EudraCT Number: 2014‐000817‐30. Comparing the effect of curcumin and genistein to treatment with Ivacaftor in CF patients with a class III mutation. clinicaltrialsregister.eu/ctr‐search/trial/2014‐000817‐30/NL (first received 22 May 2014).
EudraCT Number: 2016‐001440‐18 {published data only}
-
- EudraCT Number: 2016‐001440‐18. Pharmacokinetic interactions between ivacaftor and cytochrome P450 3A4 inhibitors in cystic fibrosis patients and healthy controls. clinicaltrialsregister.eu/ctr‐search/trial/2016‐001440‐18/NL (first received 16 January 2018).
EudraCT Number: 2016‐001619‐19 {published data only}
-
- EudraCT Number: 2016‐001619‐19. Genistein as an add‐on treatment for CF?. clinicaltrialsregister.eu/ctr‐search/trial/2016‐001619‐19/NL (first received 27 March 2016).
EUudraCT Number: 2016‐001785‐29 {published data only}
-
- EudraCT Number: 2016‐001785‐29. Combined effect of CFTR modifiers and intensive antibiotic treatment. clinicaltrialsregister.eu/ctr‐search/trial/2016‐001785‐29/IE (first received 10 May 2016).
FLAMINGO 2017 {published data only}
-
- Ent KC, Minic P, Verhulst S, Braeckel E, Flume P, Boas S, et al. GLPG2222 in subjects with cystic fibrosis homozygous for F508del: results from a phase II study (FLAMINGO). Journal of Cystic Fibrosis 2018;17(Suppl 3):S42. [CFGD Register: BD254a]
-
- Ent KC, Minic P, Verhulst S, Braeckel E, Flume P, Boas S, et al. Glpg2222 in CF subjects homozygous for f508del: results from a phase ii study (Flamingo). Pediatric Pulmonology 2018;53(S2):250. [CFGD Register: BD254b]
Horsley 2018 {published data only}
-
- Horsley A, Burr L, Kotsimbos T, Ledson M, Schwarz C, Simmonds N, et al. Safety, pharmacokinetics and pharmacodynamics of the CFTR corrector FDL169. Journal of Cystic Fibrosis 2018;17(Suppl 3):S42. [CFGD Register: BD250a]
-
- Horsley AR, Blaas S, Burr L, Carroll M, Downey DG, Drevinek P, et al. Novel CFTR corrector FDL169: safety, pharmacokinetics and pharmacodynamics. Pediatric Pulmonology 2018;53(S2):252. [CFGD Register: BD250b]
Hubert 2018 {published data only}
-
- Hubert D, Dehillotte C, Munck A, David V, Baek J, Mely L, et al. Retrospective observational study of French patients with cystic fibrosis and a Gly551Asp‐CFTR mutation after 1 and 2 years of treatment with ivacaftor in a real‐world setting. Journal of Cystic Fibrosis 2018;17(1):89‐95. [PUBMED: 28711222] - PubMed
Kerem 2014 {published data only}
-
- Ajayi T, Konstan M, Accurso FJ, Boeck K, Kerem E, Rowe S, et al. The use of high resolution computerized tomography of the chest in evaluating the effect of ataluren in nonsense mutation cystic fibrosis (nmCF) lung disease. Journal of Cystic Fibrosis 2013;12 Suppl 1:S64. [Abstract no.: 63; CENTRAL: 921666; CFGD Register: BD167h; CRS: 5500100000011670]
-
- Davies JC, Tiddens HAWM, Malfroot A, Heijerman HGM, Kerem E, Hjelte L, Sun J, et al. Ataluren in nonsense mutation cystic fibrosis patients not receiving tobramycin: significant lung function benefits in the paediatric age range. Journal of Cystic Fibrosis 2016;15 Suppl 1:S21. [Abstract no.: WS13.11; CFGD Register: BD167q]
-
- Boeck K, Heijerman HGM, Davies JC, Sermet‐Gaudelus I, Hjelte L, Kerem E, et al. Ataluren significantly reduces exacerbations in nonsense mutation cystic fibrosis patients not receiving tobramycin. Journal of Cystic Fibrosis 2016;15 Suppl 1:S20. [Abstract no.: WS13.1; CFGD Register: BD167p]
-
- Kerem E, Konstan MW, Boeck K, Accurso FJ, Sermet‐Gaudelus I, Wilschanski M, et al. Ataluren for the treatment of nonsense‐mutation cystic fibrosis: a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet Respiratory Medicine 2014;2(7):539‐47. [DOI: 10.1016/S2213-2600(14)70100-6; PUBMED: 24836205] - DOI - PMC - PubMed
-
- Kerem E, Sermet‐Gaudelus I, Hjelte L, Boeck K. Natural history of patients with cystic fibrosis carrying nonsense mutations: an analysis of placebo‐treated patients from the 009 study. Journal of cystic fibrosis 2016;15 Suppl 1:S118. [Abstract no.: 264; CFGD Register: BD167o]
KONNECTION 2013 {published data only}
-
- Boeck K, Munck A, Walker S, Faro A, Hiatt P, Chan J, et al. The effect of ivacaftor, a CFTR potentiator, in patients with cystic fibrosis and a non‐G551D‐CFTR gating mutation, the KONNECTION study. Journal of Cystic Fibrosis 2014;13 Suppl 2:S1. [Abstract no.: WS1.1; CENTRAL: 998097; CFGD Register: BD201b; CRS: 5500125000000707]
-
- Boeck K, Paskavitz J, Chen X, Higgins M. Ivacaftor, a CFTR potentiator, in cystic fibrosis patients who have a non‐G551D‐CFTR gating mutation: phase 3, part 1 results. Pediatric Pulmonology 2013;48 Suppl 36:292. [Abstract no.: 241; CENTRAL: 962356; CFGD Register: BD201a; CRS: 5500125000000421]
McCarty 2002 {published data only}
-
- Ahrens RC, Standaert TA, Launspach J, Han SH, Teresi ME, Aitken ML, et al. Use of nasal potential difference and sweat chloride as outcome measures in multicenter clinical trials in subjects with cystic fibrosis. Pediatric Pulmonology 2002;33(2):142‐50. [CFGD Register: BD136d] - PubMed
-
- Aitken ML, Ahrens RC, Karlin DA, Konstan MW, McNamara SC, Regelman WE, et al. Safety of a phase I double‐blind placebo‐controlled dose escalation trial of oral CPX in adult CF patients. Pediatric Pulmonology 1998;26(S17):276. [CFGD Register: BD136b]
-
- McCarty NA, Standaert TA, Teresi M, Tuthill C, Launspach J, Kelley TJ, et al. A phase I randomized, multicenter trial of CPX in adult subjects with mild cystic fibrosis. Pediatric Pulmonology 2002;33(2):90‐8. [CFGD Register: BD136c] - PubMed
-
- McCarty NA, Weatherly MR, Kelley TJ, Konstan MW, Milgram LJH, Teresi M, et al. Multicenter phase I trial of CPX in adults patients with mild CF: results of nasal potential difference measurements. Pediatric Pulmonology 1998;Suppl 17:276. [CFGD Register: BD136a]
McGarry 2015 {published data only}
-
- McGarry ME, Finkbeiner WE, Illek B, Fischer H, Zlock LT, Olshansky S, et al. Ivacaftor response is not predicted by signs of residual CFTR function. Pediatric Pulmonology 2015;50 Suppl 41:292. [Abstract no.: 266; CENTRAL: 1092198; CFGD Register: BD224a; CRS: 5500135000001387]
-
- Nielson DW, Moreno C, McGarry ME, Ly N. Sweat chloride as an outcome measure in n‐of‐one CF drug testing. Pediatric Pulmonology 2015;50 Suppl 41:298. [Abstract no.: 284; CENTRAL: 1092201; CFGD Register: BD224b; CRS: 5500135000001390]
NCT01549314 {published data only}
-
- NCT01549314. Cystic Fibrosis Related Bone Disease: the Role of CFTR. clinicaltrials.gov/ct2/show/NCT01549314 (first received 9 March 2012).
NCT01685801 {published data only}
-
- NCT01685801. Pilot study testing the effect of ivacaftor on lung function in subjects with cystic fibrosis and residual CFTR function [A pilot study testing the effect of ivacaftor on lung function in subjects with cystic fibrosis, residual CFTR function, and FEV1 ≥40% predicted]. clinicaltrials.gov/ct2/show/NCT01685801 (first received 14 September 2012).
NCT01784419 {published data only}
-
- NCT01784419. Short term effects of ivacaftor in non‐G551D cystic fibrosis patients. clinicaltrials.gov/ct2/show/NCT01784419 (first received 05 February 2013).
NCT01863238 {published data only}
-
- NCT01863238. An ocular safety study of ivacaftor‐treated pediatric patients 11 years of age or younger with cystic fibrosis. clinicaltrials.gov/ct2/show/NCT01863238 (first received 27 May 2013).
NCT01946412 {published data only}
-
- NCT01946412. Roll‐over study of ivacaftor in cystic fibrosis pediatric subjects with a CF transmembrane conductance regulator gene (CFTR) gating mutation. clinicaltrials.gov/ct2/show/NCT01946412 (first received 19 September 2013).
NCT02039986 {published data only}
-
- NCT02039986. Ivacaftor (Kalydeco) and insulin in cystic fibrosis (CF) [Effects of ivacaftor (Kalydeco) treatment upon insulin and incretin secretion in patients with cystic fibrosis]. clinicaltrials.gov/ct2/show/NCT02039986 (first received 20 January 2014).
NCT02141464 {published data only}
-
- NCT02141464. Energy balance and weight gain with ivacaftor treatment. clinicaltrials.gov/ct2/show/NCT02141464 (first received 19 May 2014).
NCT02310789 {published data only}
-
- NCT02310789. Does ivacaftor alter wild type CFTR‐open probability in the sweat gland secretory coil?. clinicaltrials.gov/ct2/show/NCT02310789 (first received 8 December 2014).
NCT02311140 {published data only}
-
- NCT02311140. Effects of Kalydeco on upper airway and paranasal sinus inflammation measured by nasal lavage and on symptoms (KPNSI). clinicaltrials.gov/ct2/show/NCT02311140 (first received 8 December 2014).
NCT02323100 {published data only}
-
- NCT02323100. Glycerol phenylbutyrate corrector therapy for CF (Cystic Fibrosis). clinicaltrials.gov/show/nct02323100 (first posted 23 December 2014).
NCT02443688 {published data only}
-
- NCT02443688. A phase 2 study to evaluate the efficacy, safety, and tolerability of CTX‐4430 in adult CF patients. clinicaltrials.gov/ct2/show/NCT02443688 (first received 14 May 2015).
NCT02445053 {published data only}
-
- NCT02445053. Observational study of outcomes in cystic fibrosis patients with selected gating mutations on a CFTR allele (The VOCAL Study) (VOCAL). clinicaltrials.gov/ct2/show/NCT02445053 (first received 15 May 2015).
NCT02690519 {published data only}
-
- NCT02690519. Study of GLPG1837 in subjects with cystic fibrosis (S1251N mutation) (SAPHIRA2). clinicaltrials.gov/ct2/show/NCT02690519 (first received 24 February 2016).
NCT02707562 {published data only}
-
- NCT02707562. Study of GLPG1837 in subjects with cystic fibrosis (G551D Mutation) (SAPHIRA1). clinicaltrials.gov/ct2/show/NCT02707562 (first received 14 March 2016).
NCT02709109 {published data only}
-
- NCT02709109. A study to evaluate the safety and efficacy of VX‐371 in subjects with cystic fibrosis who are homozygous for the F508del‐CFTR mutation. clinicaltrials.gov/ct2/show/NCT02709109 (first received 15 March 2016).
NCT02718495 {published data only}
-
- NCT02718495. Study assessing PTI‐428 safety, tolerability, and pharmacokinetics in subjects with cystic fibrosis. clinicaltrials.gov/ct2/show/NCT02718495 (first received 24 March 2016).
NCT02722057 {published data only}
-
- NCT02722057. A study to confirm the long‐term safety and effectiveness of Kalydeco in patients with cystic fibrosis who have an R117H‐CFTR mutation, including pediatric patients. clinicaltrials.gov/ct2/show/NCT02722057 (first received 29 March 2016).
NCT02724527 {published data only}
-
- NCT02724527. Study of cavosonstat (N91115) in CF patients who are heterozygous for F508del‐CFTR and a gating mutation and being treated with ivacaftor (SNO‐7). clinicaltrials.gov/ct2/show/NCT02724527 (first received 31 March 2016).
NCT02742519 {published data only}
-
- NCT02742519. A study to evaluate efficacy and safety of ivacaftor in subjects with cystic fibrosis aged 3 through 5 years who have a specified CFTR gating mutation. clinicaltrials.gov/ct2/show/NCT02742519 (first received 19 April 2015).
NCT02759562 {published data only}
-
- NCT02759562. Effect of andecaliximab on FEV1 in adults with cystic fibrosis. clinicaltrials.gov/ct2/show/NCT02759562 (first received 03 May 2016).
NCT02934698 {published data only}
-
- NCT02934698. An efficacy and safety study of ivacaftor in patients with cystic fibrosis and two splicing mutations. clinicaltrials.gov/ct2/show/NCT02934698 (first received 17 October 2016).
NCT03068312 {published data only}
-
- NCT03068312. A study to evaluate efficacy of ivacaftor in subjects with cystic fibrosis who have a 3849 + 10KB C→T or D1152H CFTR mutation. clinicaltrials.gov/ct2/show/NCT03068312 (first received 01 March 2017).
NCT03256799 {published data only}
-
- NCT03256799. Evaluation of ivacaftor in patients using ataluren for nonsense mutations. clinicaltrials.gov/ct2/show/NCT03256799 (first received 22 August 2017).
NCT03256968 {published data only}
-
- NCT03256968. PTC study to evaluate ataluren in combination with ivacaftor. clinicaltrials.gov/ct2/show/NCT03256968 (first received 22 August 2017).
NCT03258424 {published data only}
-
- NCT03258424. Study assessing PTI‐428 safety, tolerability, and pharmacokinetics in subjects with cystic fibrosis on KALYDECO® as background therapy. clinicaltrials.gov/ct2/show/NCT03258424 (first received 23 August 2017).
NCT03277196 {published data only}
-
- NCT03277196. A phase 3, 2‐arm, open‐label study to evaluate the safety and pharmacodynamics of long‐term ivacaftor treatment in subjects with cystic fibrosis who are less than 24 months of age at treatment initiation and have an approved ivacaftor‐responsive mutation. clinicaltrials.gov/ct2/show/NCT03277196 (first received 08 September 2017).
NCT03390985 {published data only}
-
- NCT03390985. Canadian observation trial in CF patients undergoing treatment with ivacaftor (G551D). clinicaltrials.gov/ct2/show/NCT03390985 (first received 5 January 2018).
NCT03474042 {published data only}
-
- NCT03474042. GLPG2737 on top of Orkambi in subjects with cystic fibrosis (PELICAN). clinicaltrials.gov/ct2/show/NCT03474042 (first received 22 March 2018).
NCT03652090 {published data only}
-
- NCT03652090. Evaluation of the Primary Human Nasal Epithelial Cell Culture Model in the Context of Personalized Therapy in Cystic Fibrosis. clinicaltrials.gov/ct2/show/NCT03652090 (first received 29 August 2018).
PERSIST 2014 {published data only}
-
- McKone EF, Borowitz D, Drevinek P, Griese M, Konstan MW, Wainwright C, et al. Long‐term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the Gly551Asp‐CFTR mutation: a phase 3, open‐label extension study (PERSIST). The Lancet. Respiratory medicine 2014;2(11):902‐10. [PUBMED: 25311995] - PubMed
Pradal 2002 {published data only}
-
- Pradal U, Casotti V, Delmarco A, Nicolis E, Livraghi A, Conese M, et al. Effects of gentamicin on ion transport, MRNA and protein CFTR expression in patients with R1162X: A double blind placebo controlled study. Pediatric Pulmonology 2002;34 Suppl 24:263. [CFGD Register: BD145]
RIO‐CF 2017 {published data only}
-
- Derichs N, Taylor‐Cousar J, Tullis E, Davies J, Nazareth D, Downey DG, et al. Safety, tolerability and early signs of efficacy with riociguat for the treatment of adult Phe508del homozygous cystic fibrosis patients: study design and rationale for the Rio‐CF study. Journal of Cystic Fibrosis 2017;16(Supplement 1):S36. [CFGD Register: BD246b]
-
- Taylor‐Cousar JL, Derichs N, Tullis DE, Davies JC, Nazareth D, Downey D, et al. Safety, tolerability and early signs of efficacy with riociguat for the treatment of adult Phe508del homozygous cystic fibrosis patients: safety data from the rio‐CF study. Pediatric Pulmonology 2017;52(Supplement 47):307. [CFGD Register: BD246c]
-
- Taylor‐Cousar JL, Tullis E, Derichs N, Davies JC, Nazareth D, Downey D, et al. Riociguat for the treatment of adult Phe508del homozygous cystic fibrosis: efficacy data from the Phase II Rio‐CF study. Journal of Cystic Fibrosis 2018;17(Suppl 3):S67. [CFGD Register: BD246a] - PubMed
Romano 2000 {published data only}
-
- Romano L, Casciaro R, Vanini P, Zegarra‐Moran O, Negro I, Minuto N, et al. Reduction of sweat ion concentrations following topical application of gentamicin in CF patients carrying nonsense mutations. Proceedings of 24th European Cystic Fibrosis Conference; 2001 June 6‐9; Vienna, Austria. 2001:P11. [CFGD Register: BD144b]
-
- Romano L, Sacchi R, Zegarra‐Moran O, Vanini P, Guerriero F, Casciaro R, et al. Effects of topical applications of gentamicin on sweat test in CF patients carrying nonsense‐mutations. Pediatric Pulmonology 2000;30 Suppl 20:250. [CFGD Register: BD144a]
Rubenstein 1998 {published data only}
-
- Rubenstein RC, Zeitlin PL. A pilot clinical trial of oral sodium 4‐phenylbutyrate (Buphenyl) in deltaF508‐homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. American Journal of Respiratory and Critical Care Medicine 1998;157(2):484‐90. [CFGD Register: BD146b; MEDLINE: ] - PubMed
-
- Rubenstein RC, Zeitlin PL. A randomized, double blind, placebo‐controlled trial of sodium 4‐phenylbutyrate (Buphenyl) in deltaF508‐homozygous cystic fibrosis patients: Partial restoration of nasal epithelial CFTR function. Pediatric Pulmonology 1997;24 Suppl 14:272. [CFGD Register: BD146a] - PubMed
Rubenstein 2006 {published data only}
-
- Rubenstein RC, Propert KJ, Reenstra WW, Skotleski ML. A pilot trial of the combination of phenylbutyrate and genistein. Pediatric Pulmonology 2006;41 Suppl 29:294. [CFGD Register: BD149]
Seliger 2015 {published data only}
-
- Seliger V, Bai Y, Volkova N, Tian S, Waltz. Prevalance of cataracts in a population of cystic fibrosis patients homozygous for the F508del mutation. Journal of Cystic Fibrosis 2015;14 Suppl 1:S108. [Abstract no.: 196; CFGD Register: BD213d // BD214c]
Sermet‐Gaudelus 2010 {published data only}
-
- Sermet‐Gaudelus I, Boeck K, Casimir GJ, Vermeulen F, Leal T, Mogenet A, et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. American Journal of Respiratory & Critical Care Medicine 2010;182(10):1262‐72. [CFGD Register: BD167b; MEDLINE: ] - PubMed
TOPIC 2018 {published data only}
-
- NCT03085485. The Topic Trial ‐ Study to Determine the Safety and Efficacy of Ivacaftor. clinicaltrials.gov/ct2/show/NCT03085485 (first received 21 March 2017).
Wilschanski 2003 {published data only}
-
- Wilschanski M, Virgilis D, Strauss‐Liviatan N, Tal A, Bentur L, Blau H, et al. Gentamicin causes functional expression of CFTR in CF patients carrying stop mutations: a double‐blind placebo controlled trial. Pediatric Pulmonology 2000;30 Suppl 20:244. [CFGD Register: BD143a; MEDLINE: ]
-
- Wilschanski M, Yahav J, Blau H, Bentur L, Rivlin J, Aviram M, et al. Restoration of CFTR function by gentamicin in cystic fibrosis patients carrying stop mutations: a double blind placebo controlled trial. Gastroenterology 2003;124(4 Suppl 1):A582. [CFGD Register: BD143c]
-
- Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, et al. Gentamicin‐induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. New England Journal of Medicine 2003;349(15):1433‐41. [CFGD Register: BD143b] - PubMed
Wilschanski 2008 {published data only}
-
- Kerem E, Yaakov Y, Armoni S, Pugatsch T, Shoseyov D, Cohen M, et al. PTC124 induces time‐dependent improvements in chloride conductance and clinical parameters in patients with nonsense‐mutation‐mediated cystic fibrosis. Pediatric Pulmonology 2008;43 Suppl 31:294. [CFGD Register: BD22d; MEDLINE: ]
-
- Sermet‐Gaudelus I, Boeck K, Casimir G, Leal T, Vermeulen F, Mogenet A, et al. Children with nonsense‐mutation‐mediated cystic fibrosis respond to investigational treatment with PTC124. Pediatric Pulmonology 2008;43 Suppl 31:313. [CFGD Register: BD22c; MEDLINE: ]
-
- Sermet‐Gaudelus I, Leal T, Boeck K, Casimir G, Hanssens L, Hage P, et al. PTC124 induces CFTR full‐length production and activity in children with nonsense‐mutation‐mediated CF. Journal of Cystic Fibrosis 2008;7 Suppl 2:S22. [CFGD Register: BD22a; MEDLINE: ]
-
- Wilschanski M, Armoni S, Yaakov Y, Blau H, Shoseyov D, Cohen M, et al. PTC124 treatment over 3 months improves pharmacodynamic and clinical parameters in patients with nonsense ‐mutation‐mediated CF. Journal of Cystic Fibrosis 2008;7 Suppl 2:S22. [CFGD Register: BD22b; MEDLINE: ]
Zeitlin 2002 {published data only}
-
- Zeitlin PL, Diener‐West M, Rubenstein RC, Boyle MP, Lee CK, Brass‐Ernst L. Evidence of CFTR function in cystic fibrosis after systemic administration of 4‐phenylbutyrate. Molecular Therapy 2002;6(1):119‐26. [CFGD Register: BD148] - PubMed
References to studies awaiting assessment
Kazani 2016 {published data only}
-
- Kazani S, Alcantara J, Debonnett L, Doucet J, Jones I, Kulmatycki K, et al. QBW251 is a safe and efficacious CFTR potentiator for patients with cystic fibrosis. American Journal of Respiratory and Critical Care Medicine 2016;193(Meeting Abstracts):A7789. [CFGD Register: BD243]
-
- NCT02190604. Safety, Tolerability, Pharmacokinetics, and Preliminary Pharmacodynamics of QBW251 in Healthy Subjects and Cystic Fibrosis Patients.. clinicaltrials.gov/ct2/show/NCT02190604 (first received 15 July 2014).
Uttamsingh 2016 {published data only}
-
- Uttamsingh V, Pilja L, Brummel CL, Grotbeck B, Cassella JV, Braman G. CTP‐656 multiple dose pharmacokinetic profile continues to support a once‐daily potentiator for cystic fibrosis patients with gating mutations. Pediatric Pulmonology 2016;51 Suppl 45:277. [Abstract no.: 244; CFGD Register: BD241]
Additional references
Amaral 2007
-
- Amaral MD, Kunzelmann K. Molecular targeting of CFTR as a therapeutic approach to cystic fibrosis. Trends in Pharmacological Sciences 2007;28(7):334‐41. - PubMed
Aslam 2017
Bobadilla 2002
-
- Bobadilla JL, Macek M, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations‐‐correlation with incidence data and application to screening. Human Mutation 2002;19(6):575‐606. - PubMed
CFMD 2012
-
- Hospital for Sick Children in Toronto. Cystic Fibrosis Mutation Database. www.genet.sickkids.on.ca/StatisticsPage.html (accessed 01 March 2012).
Deeks 2011
-
- Deeks J, Higgins J, Altman D. Chapter 9 Analysing data and undertaking meta‐analysis. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hamosh 1998
-
- Hamosh A, FitzSimmons SC, Macek Jr M, Knowles MR, Rosenstein BJ, Cutting GR. Comparison of the clinical manifestations of cystic fibrosis in black and white patients. Journal of Pediatrics 1998;132(2):255‐9. - PubMed
Higgins 2003
Higgins 2011a
-
- Higgins JPT, Altman DG (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011b
-
- Higgins JPT, Deeks JJ, Altman DG on behalf of the Cochrane Statistical Methods Group. Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors). Cochrane Handbook of Systematic Reviews of Interventions. Version 5.1 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
KONTINUE 2017
-
- NCT01707290. Rollover study of ivacaftor in subjects with cystic fibrosis and a non G551D CFTR mutation (KONTINUE) [A phase 3, two‐arm, rollover study to evaluate the safety of long term ivacaftor treatment in subjects 6 years of age and older with cystic fibrosis and a non‐G551D CFTR mutation]. clinicaltrials.gov/ct2/show/NCT01707290 (first received 16 October 2012).
McKone 2004
-
- McKone EF, Aitken ML. Cystic fibrosis: disease mechanisms and therapeutic targets. Drug Discovery Today: Disease Mechanisms 2004;1(1):137‐43.
Quittner 2009
-
- Quittner AL, Modi AC, Wainwright C, Otto K, Kirihara J, Montgomery AB. Determination of the minimal clinically important difference scores for the Cystic Fibrosis Questionnaire‐Revised respiratory symptom scale in two populations of patients with cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. Chest 2009;135(6):1610‐8. - PMC - PubMed
Riordan 1989
-
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989;245(4922):1066. - PubMed
Rowntree 2003
-
- Rowntree RK, Harris A. The phenotypic consequences of CFTR mutations. Annals of Human Genetics 2003;67(5):471‐85. - PubMed
Southern 1997
Southern 2007
-
- Southern KW. Cystic fibrosis and formes frustes of CFTR‐related disease. Respiration: International Review of Thoracic Diseases 2007;74(3):241‐51. - PubMed
Southern 2018
UK CF Registry Report 2013
-
- UK CF Trust. Cystic fibrosis: our focus. www.cysticfibrosis.org.uk/media/598466/annual‐data‐report‐2013‐jul14.pdf (accessed 07 Jan 2015).
Van Goor 2009
-
- Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX‐770.. Proceedings of the National Academy of Sciences of the United States of America 2009;106(44):18825‐30. [DOI: 10.1073/pnas.0904709106] - DOI - PMC - PubMed
Whiting 2014
-
- Whiting P, Al M, Burgers L, Westwood M, Ryder S, Hoogendoorn M, Armstrong N, et al. Ivacaftor for the treatment of patients with cystic fibrosis and the G551D mutation: a systematic review and cost‐effectiveness analysis. Health Technology Assessment (Winchester, England) 2014;18(18):1‐106. [DOI: 10.3310/hta18180] - DOI - PMC - PubMed
References to other published versions of this review
Patel 2012
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical