

UTRme: A Scoring-Based Tool to Annotate Untranslated Regions in Trypanosomatid Genomes
- PMID: 30619487
- PMCID: PMC6305552
- DOI: 10.3389/fgene.2018.00671
UTRme: A Scoring-Based Tool to Annotate Untranslated Regions in Trypanosomatid Genomes
Abstract
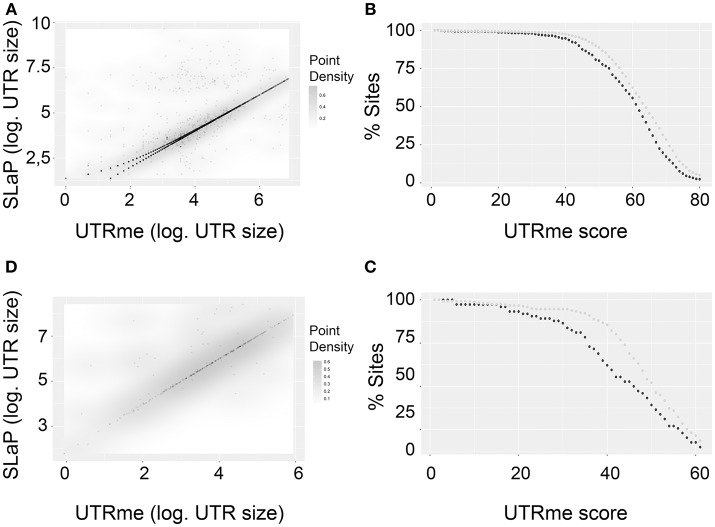
Most signals involved in post-transcriptional regulatory networks are located in the untranslated regions (UTRs) of the mRNAs. Therefore, to deepen our understanding of gene expression regulation, delimitation of these regions with high accuracy is needed. The trypanosomatid lineage includes a variety of parasitic protozoans causing a significant worldwide burden on human health. Given their peculiar mechanisms of gene expression, these organisms depend on post-transcriptional regulation as the main level of gene expression control. In this context, the definition of the UTR regions becomes of key importance. We have developed UTR-mini-exon (UTRme), a graphical user interface (GUI) stand-alone application to identify and annotate 5' and 3' UTR regions in a highly accurate way. UTRme implements a multiple scoring system tailored to address the issue of false positive UTR assignment that frequently arise because of the characteristics of the intergenic regions. Even though it was developed for trypanosomatids, the tool can be used to predict 3' sites in any eukaryote and 5' UTRs in any organism where trans-splicing occurs (such as the model organism C. elegans). UTRme offers a way for non-bioinformaticians to precisely determine UTRs from transcriptomic data. The tool is freely available via the conda and github repositories.
Keywords: GUI; UTR prediction software; post transcriptional regulation; prediction score; untranslated region.
Figures
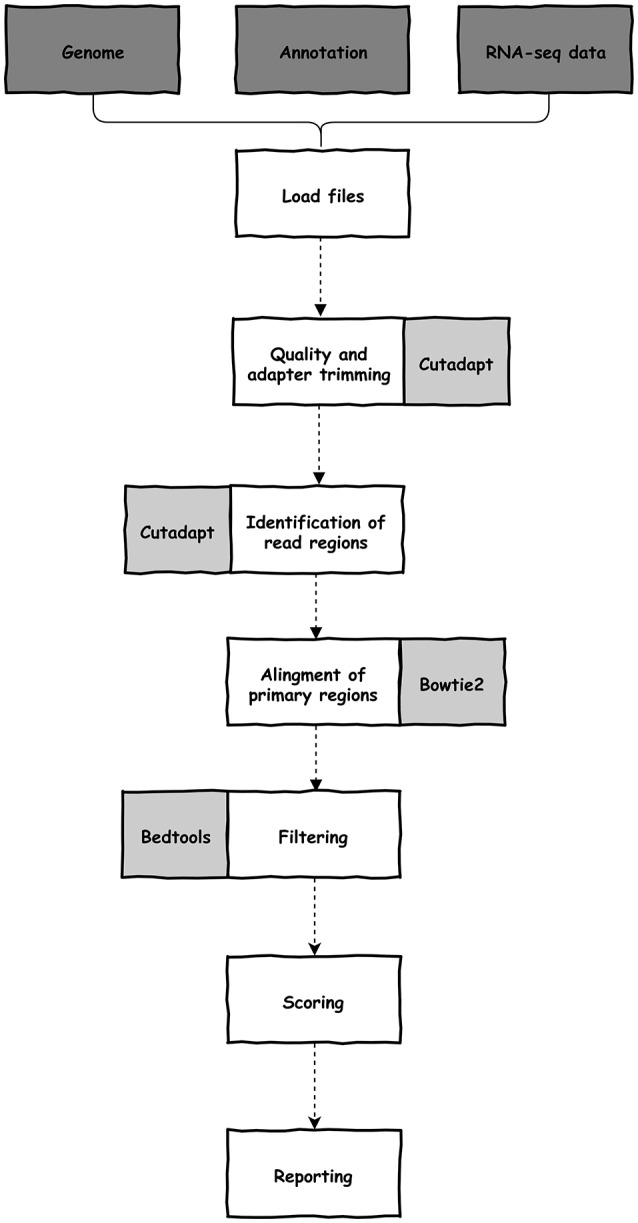

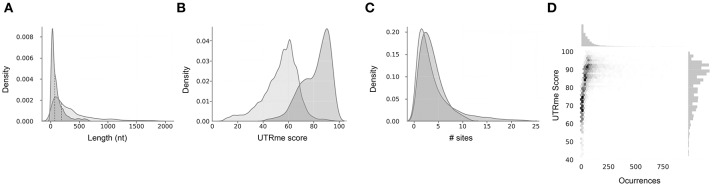
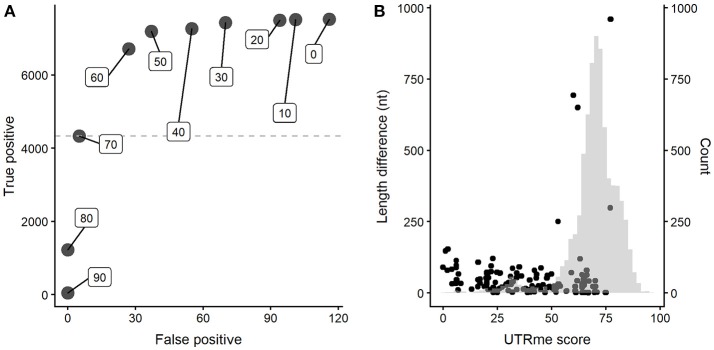
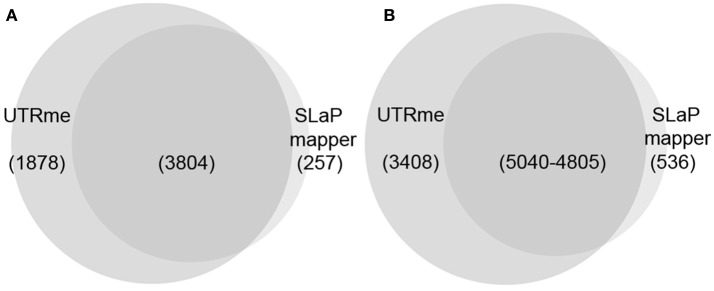
References
-
- Bontempi E. J., Porcel B. M., Henriksson J., Carlsson L., Rydaker M., Segura E. L., et al. (1994). Genes for histone H3 in Trypanosoma cruzi. Mol. Biochem. Parasitol. 66, 147–151. - PubMed
LinkOut - more resources
Full Text Sources