

DEGS1-associated aberrant sphingolipid metabolism impairs nervous system function in humans
- PMID: 30620338
- PMCID: PMC6391115
- DOI: 10.1172/JCI124159
DEGS1-associated aberrant sphingolipid metabolism impairs nervous system function in humans
Abstract
Background: Sphingolipids are important components of cellular membranes and functionally associated with fundamental processes such as cell differentiation, neuronal signaling, and myelin sheath formation. Defects in the synthesis or degradation of sphingolipids leads to various neurological pathologies; however, the entire spectrum of sphingolipid metabolism disorders remains elusive.
Methods: A combined approach of genomics and lipidomics was applied to identify and characterize a human sphingolipid metabolism disorder.
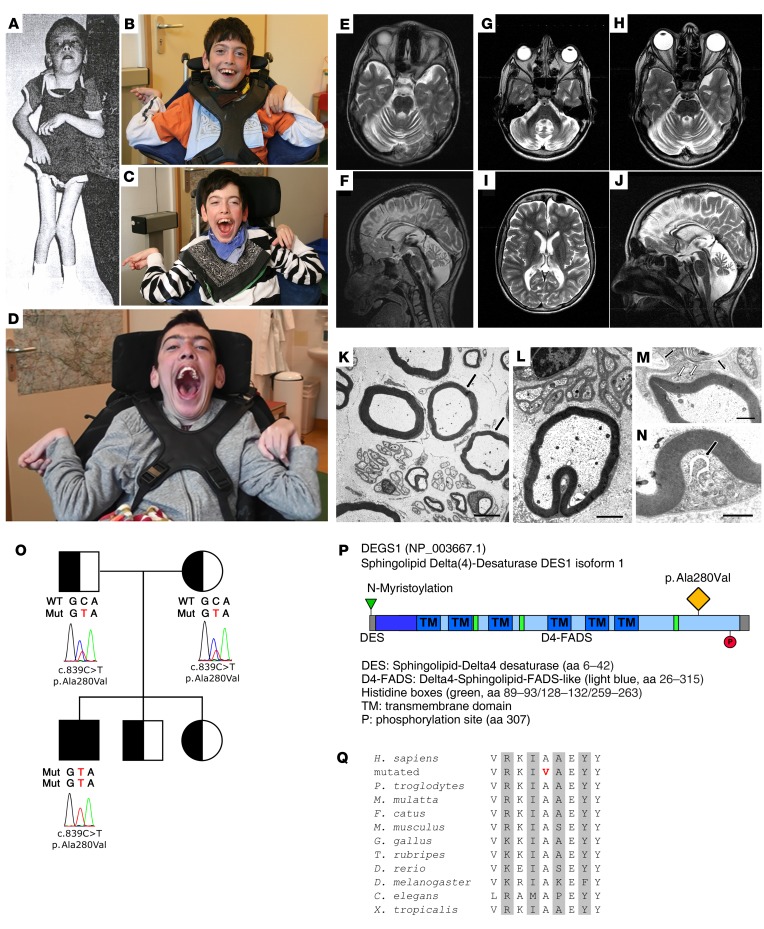
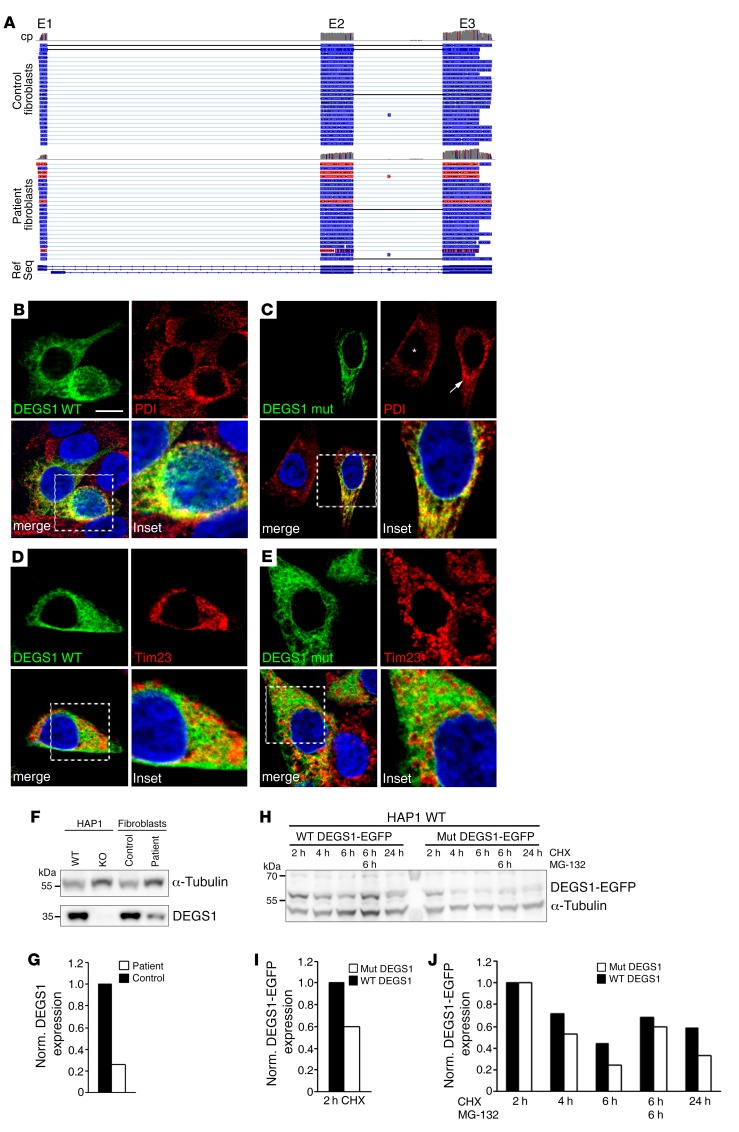
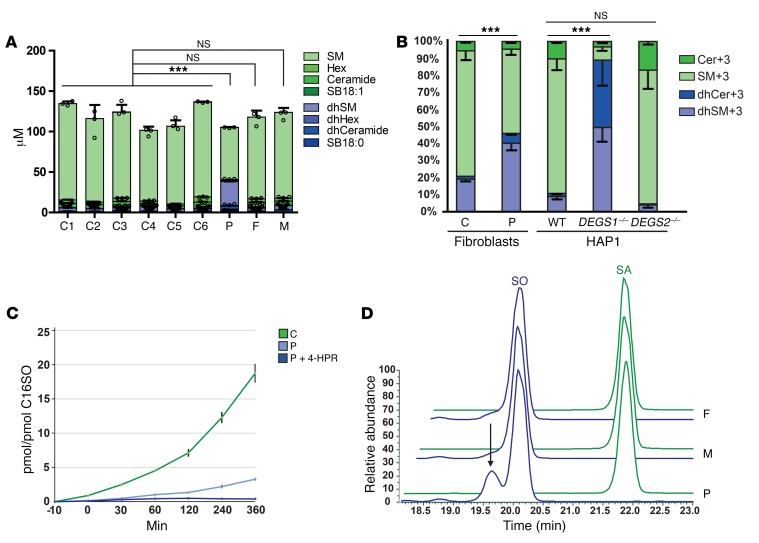
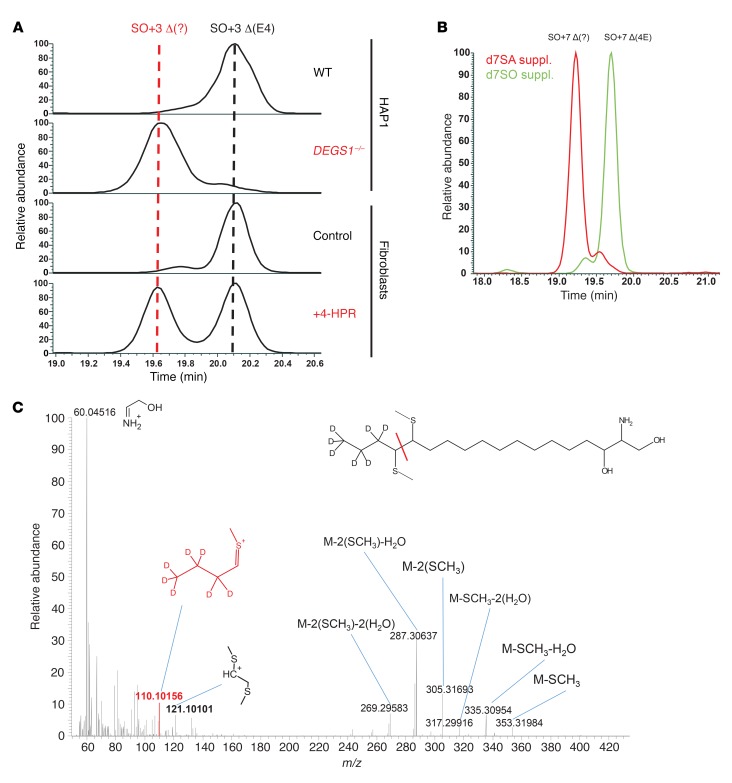
Results: By whole-exome sequencing in a patient with a multisystem neurological disorder of both the central and peripheral nervous systems, we identified a homozygous p.Ala280Val variant in DEGS1, which catalyzes the last step in the ceramide synthesis pathway. The blood sphingolipid profile in the patient showed a significant increase in dihydro sphingolipid species that was further recapitulated in patient-derived fibroblasts, in CRISPR/Cas9-derived DEGS1-knockout cells, and by pharmacological inhibition of DEGS1. The enzymatic activity in patient fibroblasts was reduced by 80% compared with wild-type cells, which was in line with a reduced expression of mutant DEGS1 protein. Moreover, an atypical and potentially neurotoxic sphingosine isomer was identified in patient plasma and in cells expressing mutant DEGS1.
Conclusion: We report DEGS1 dysfunction as the cause of a sphingolipid disorder with hypomyelination and degeneration of both the central and peripheral nervous systems.
Trial registration: Not applicable.
Funding: Seventh Framework Program of the European Commission, Swiss National Foundation, Rare Disease Initiative Zurich.
Keywords: Demyelinating disorders; Genetics; Metabolism; Monogenic diseases; Neurological disorders.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
