

Exonic CLDN16 mutations associated with familial hypomagnesemia with hypercalciuria and nephrocalcinosis can induce deleterious mRNA alterations
- PMID: 30621608
- PMCID: PMC6325764
- DOI: 10.1186/s12881-018-0713-7
Exonic CLDN16 mutations associated with familial hypomagnesemia with hypercalciuria and nephrocalcinosis can induce deleterious mRNA alterations
Abstract
Background: Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis type 1 is an autosomal recessive disease characterized by excessive renal magnesium and calcium excretion, bilateral nephrocalcinosis, and progressive chronic renal failure. This rare disease is caused by mutations in CLDN16 that encodes claudin-16, a tight-junction protein involved in paracellular reabsorption of magnesium and calcium in the renal tubule. Most of these variants are located in exons and have been classified as missense mutations. The functional consequences of some of these claudin-16 mutant proteins have been analysed after heterologous expression showing indeed a significant loss of function compared to the wild-type claudin-16. We hypothesize that a number of CLDN16 exonic mutations can be responsible for the disease phenotype by disrupting the pre-mRNA splicing process.
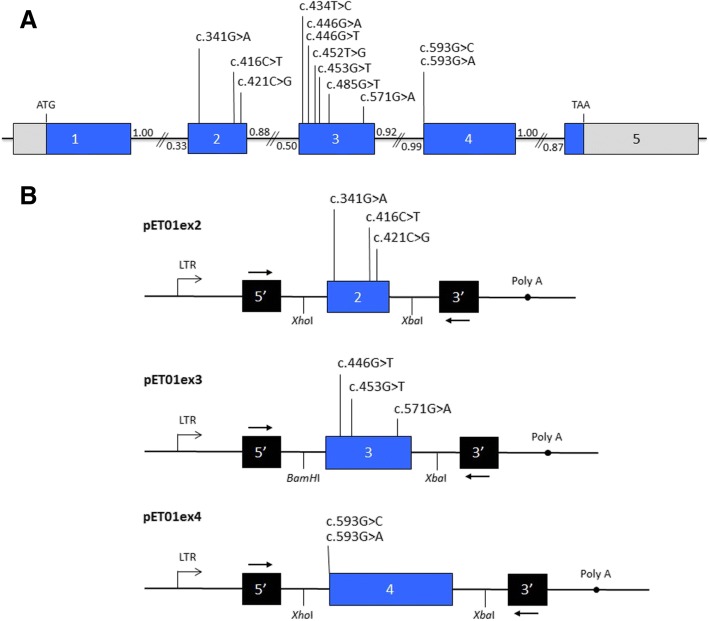
Methods: We selected 12 previously described presumed CLDN16 missense mutations and analysed their potential effect on pre-mRNA splicing using a minigene assay.
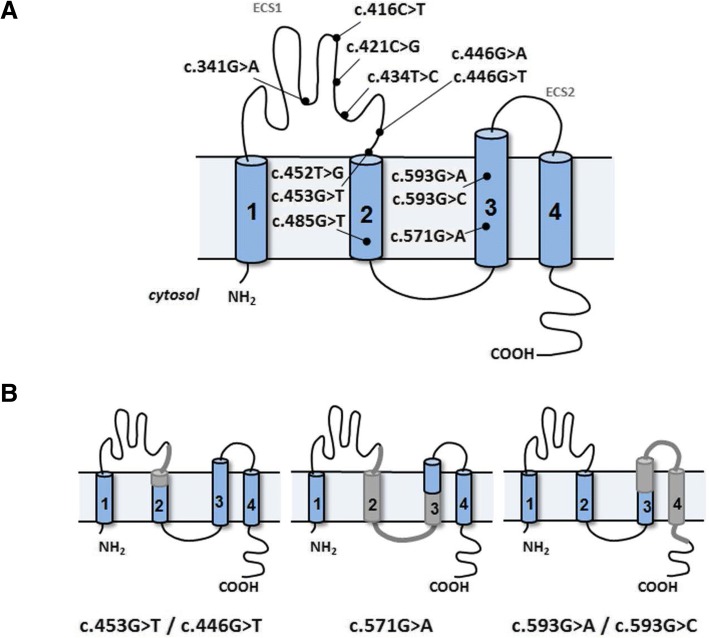
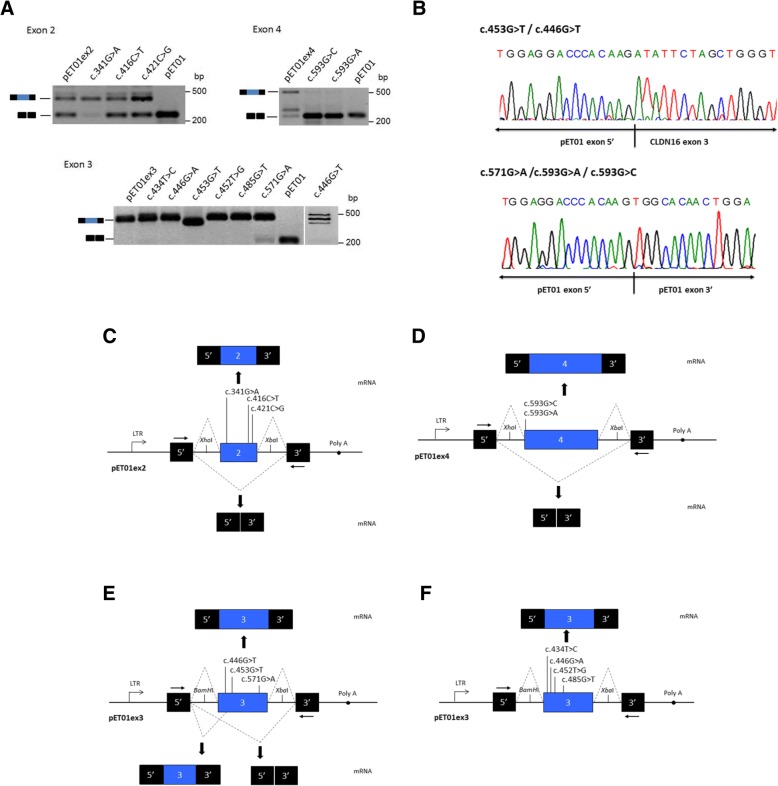
Results: Our results indicate that five of these mutations induce significant splicing alterations. Mutations c.453G > T and c.446G > T seem to inactivate exonic splicing enhancers and promote the use of an internal cryptic acceptor splice site resulting in inclusion of a truncated exon 3 in the mature mRNA. Mutation c.571G > A affects an exonic splicing enhancer resulting in partial skipping of exon 3. Mutations c.593G > C and c.593G > A disturb the acceptor splice site of intron 3 and cause complete exon 4 skipping.
Conclusions: To our knowledge, this is the first report of CLDN16 exonic mutations producing alterations in splicing. We suggest that in the absence of patients RNA samples, splicing functional assays with minigenes could be valuable for evaluating the effect of exonic CLDN16 mutations on pre-mRNA splicing.
Keywords: Bioinformatics analysis; Claudin-16; Exonic splicing enhancer; Minigene; Missense mutation; Pre-mRNA splicing; Splicing defects.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests. The funders had no role in study design, data collection and analysis, preparation of the manuscript or in the decision to publish the results.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
Characterization of two novel mutations in the claudin-16 and claudin-19 genes that cause familial hypomagnesemia with hypercalciuria and nephrocalcinosis.Gene. 2019 Mar 20;689:227-234. doi: 10.1016/j.gene.2018.12.024. Epub 2018 Dec 18. Gene. 2019. PMID: 30576809
-
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: unusual clinical associations and novel claudin16 mutation in an Egyptian family.Clin Exp Nephrol. 2009 Aug;13(4):288-294. doi: 10.1007/s10157-008-0126-6. Epub 2009 Jan 24. Clin Exp Nephrol. 2009. PMID: 19165416
-
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: phenotype-genotype correlation and outcome in 32 patients with CLDN16 or CLDN19 mutations.Clin J Am Soc Nephrol. 2012 May;7(5):801-9. doi: 10.2215/CJN.12841211. Epub 2012 Mar 15. Clin J Am Soc Nephrol. 2012. PMID: 22422540 Free PMC article.
-
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis.Pediatr Nephrol. 2021 Oct;36(10):3045-3055. doi: 10.1007/s00467-021-04968-2. Epub 2021 Feb 17. Pediatr Nephrol. 2021. PMID: 33595712 Review.
-
Claudins and mineral metabolism.Curr Opin Nephrol Hypertens. 2016 Jul;25(4):308-13. doi: 10.1097/MNH.0000000000000239. Curr Opin Nephrol Hypertens. 2016. PMID: 27191348 Free PMC article. Review.
Cited by
-
Six Exonic Variants in the SLC5A2 Gene Cause Exon Skipping in a Minigene Assay.Front Genet. 2020 Nov 5;11:585064. doi: 10.3389/fgene.2020.585064. eCollection 2020. Front Genet. 2020. PMID: 33250922 Free PMC article.
-
Characterization of pre-mRNA Splicing Defects Caused by CLCN5 and OCRL Mutations and Identification of Novel Variants Associated with Dent Disease.Biomedicines. 2023 Nov 17;11(11):3082. doi: 10.3390/biomedicines11113082. Biomedicines. 2023. PMID: 38002082 Free PMC article.
-
A Novel CLCN5 Splice Site Mutation in a Boy with Incomplete Phenotype of Dent Disease.J Pediatr Genet. 2019 Dec;8(4):235-239. doi: 10.1055/s-0039-1692172. Epub 2019 Jun 4. J Pediatr Genet. 2019. PMID: 31687264 Free PMC article.
-
Claudins in Renal Physiology and Pathology.Genes (Basel). 2020 Mar 10;11(3):290. doi: 10.3390/genes11030290. Genes (Basel). 2020. PMID: 32164158 Free PMC article. Review.
-
Identification of mutations in 15 nephrolithiasis-related genes leading to a molecular diagnosis in 85 Chinese pediatric patients.Pediatr Nephrol. 2023 Nov;38(11):3645-3661. doi: 10.1007/s00467-023-06028-3. Epub 2023 Jun 12. Pediatr Nephrol. 2023. PMID: 37306718
References
-
- Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, Vitzthum H, Suzuki Y, Luk JM, Becker C, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet. 2006;79:949–957. doi: 10.1086/508617. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
