

Emerging Pharmacological Targets for the Treatment of Nonalcoholic Fatty Liver Disease, Insulin Resistance, and Type 2 Diabetes
- PMID: 30625285
- PMCID: PMC7198260
- DOI: 10.1146/annurev-pharmtox-010716-104727
Emerging Pharmacological Targets for the Treatment of Nonalcoholic Fatty Liver Disease, Insulin Resistance, and Type 2 Diabetes
Abstract
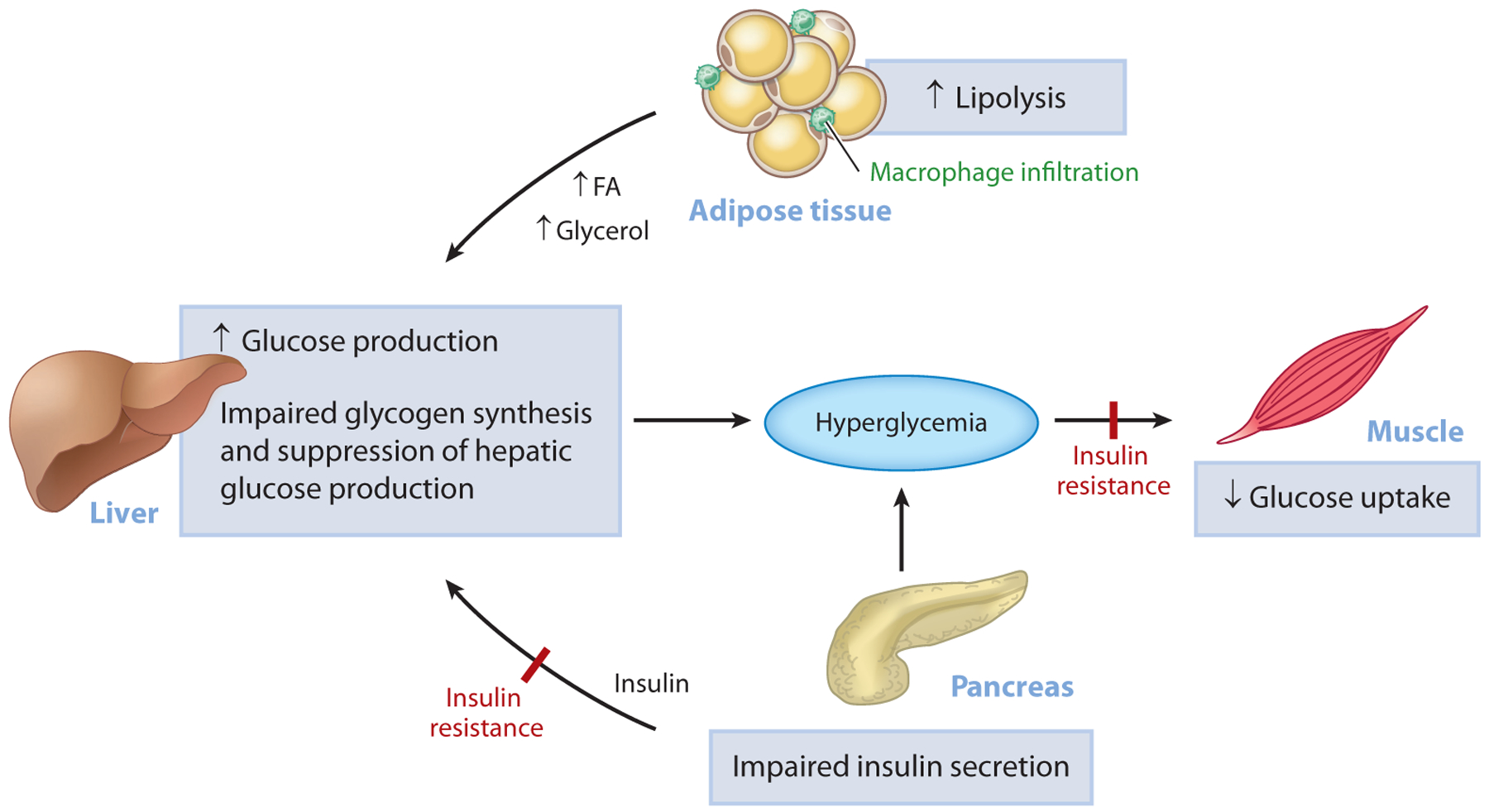
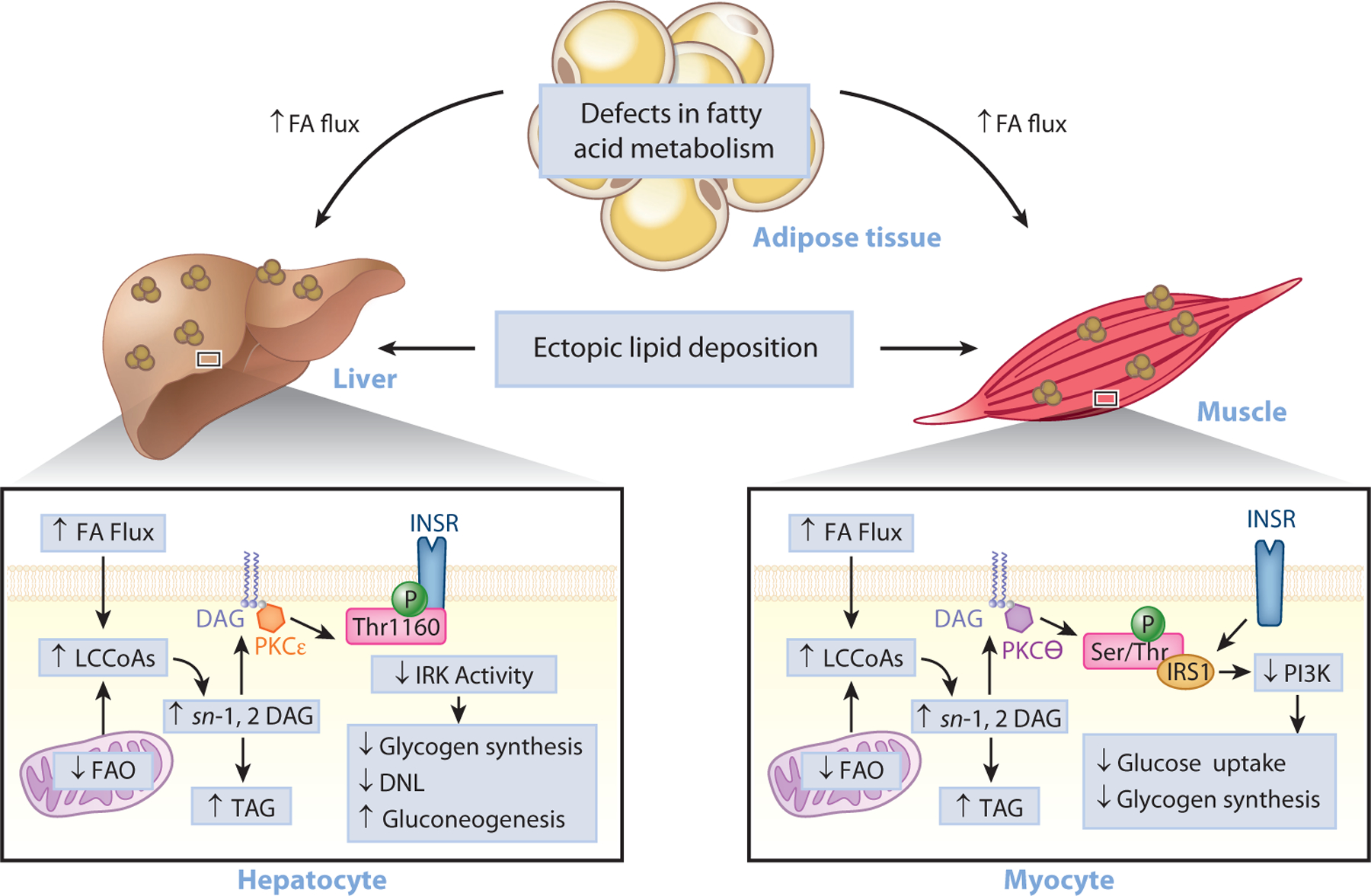
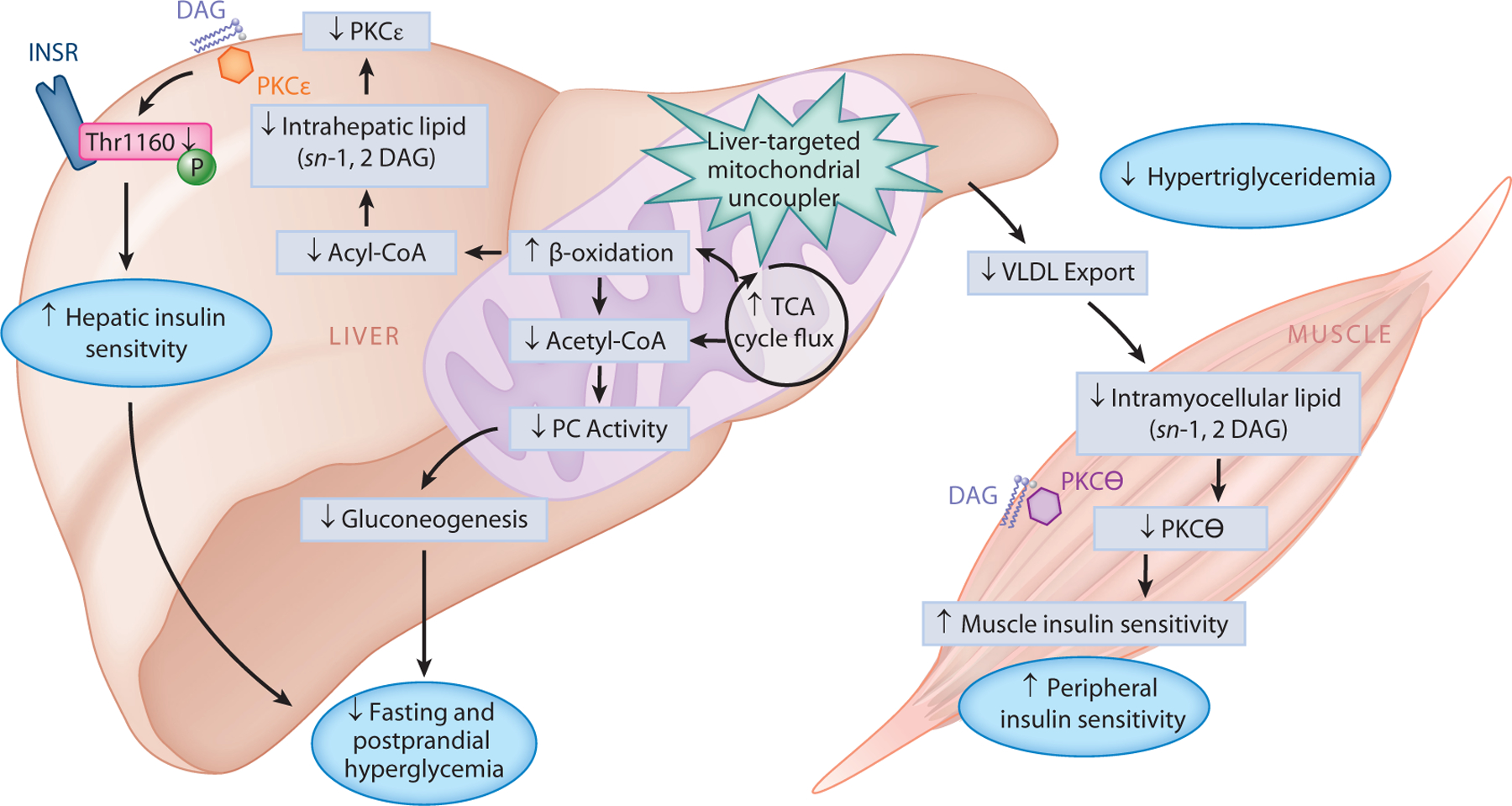
Type 2 diabetes (T2D) is characterized by persistent hyperglycemia despite hyperinsulinemia, affects more than 400 million people worldwide, and is a major cause of morbidity and mortality. Insulin resistance, of which ectopic lipid accumulation in the liver [nonalcoholic fatty liver disease (NAFLD)] and skeletal muscle is the root cause, plays a major role in the development of T2D. Although lifestyle interventions and weight loss are highly effective at reversing NAFLD and T2D, weight loss is difficult to sustain, and newer approaches aimed at treating the root cause of T2D are urgently needed. In this review, we highlight emerging pharmacological strategies aimed at improving insulin sensitivity and T2D by altering hepatic energy balance or inhibiting key enzymes involved in hepatic lipid synthesis. We also summarize recent research suggesting that liver-targeted mitochondrial uncoupling may be an attractive therapeutic approach to treat NAFLD, nonalcoholic steatohepatitis, and T2D.
Keywords: ectopic lipids; insulin resistance; liver-targeted mitochondrial uncoupling; type 2 diabetes.
Figures
References
-
- Ogurtsova K, da Rocha Fernandes JD, Huang Y, Linnenkamp U, Guariguata L, et al. 2017. IDF Diabetes Atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract 128:40–50 - PubMed
-
- DeFronzo RA, Ferrannini E, Groop L, Henry RR, Herman WH, et al. 2015. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 1:15019. - PubMed
-
- Wang Y, Beydoun MA, Liang L, Caballero B, Kumanyika SK. 2008. Will all Americans become overweight or obese? Estimating the progression and cost of the US obesity epidemic. Obesity 16:2323–30 - PubMed
-
- Boyle JP, Honeycutt AA, Narayan KM, Hoerger TJ, Geiss LS, et al. 2001. Projection of diabetes burden through 2050: impact of changing demography and disease prevalence in the U.S. Diabetes Care 24:1936–40 - PubMed
-
- Finkelstein EA, Trogdon JG, Cohen JW, Dietz W. 2009. Annual medical spending attributable to obesity: payer-and service-specific estimates. Health Aff. 28:w822–31 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
