

Challenges in Clinical Metaproteomics Highlighted by the Analysis of Acute Leukemia Patients with Gut Colonization by Multidrug-Resistant Enterobacteriaceae
- PMID: 30626002
- PMCID: PMC6473847
- DOI: 10.3390/proteomes7010002
Challenges in Clinical Metaproteomics Highlighted by the Analysis of Acute Leukemia Patients with Gut Colonization by Multidrug-Resistant Enterobacteriaceae
Abstract
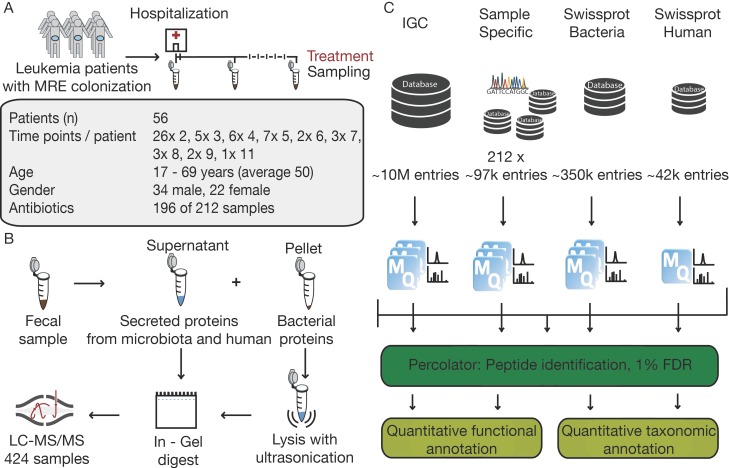
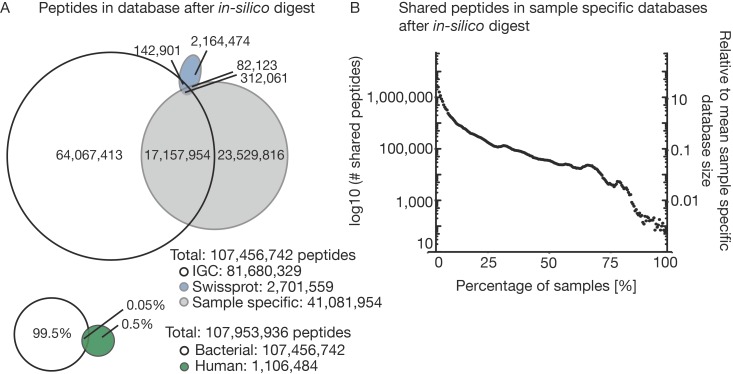
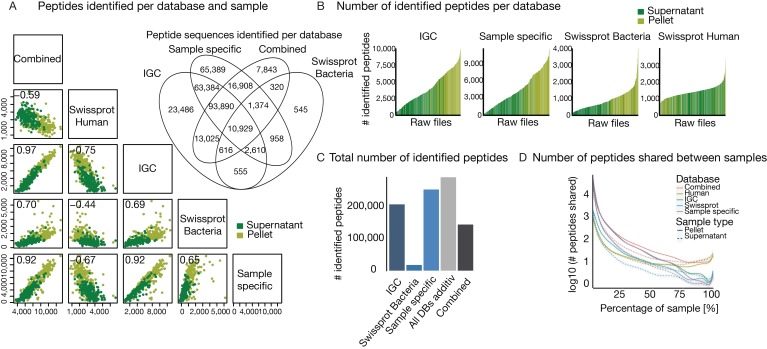
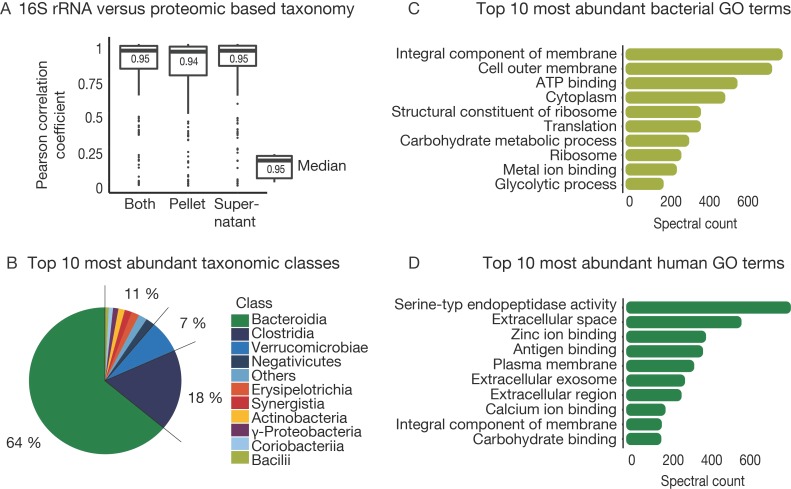
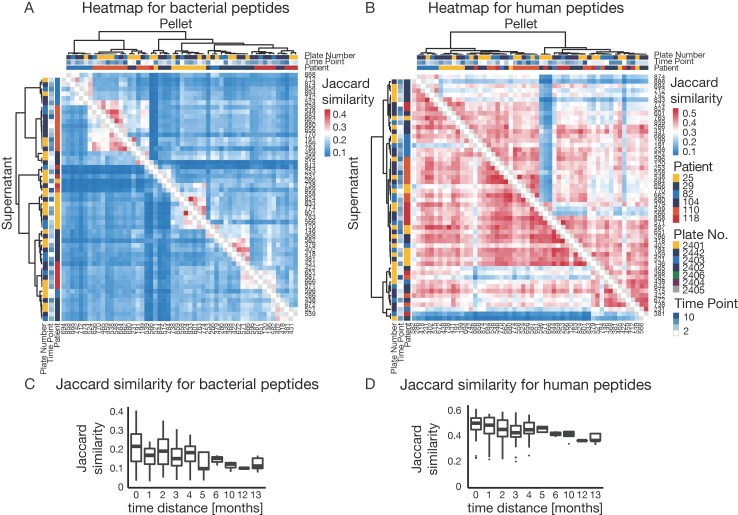
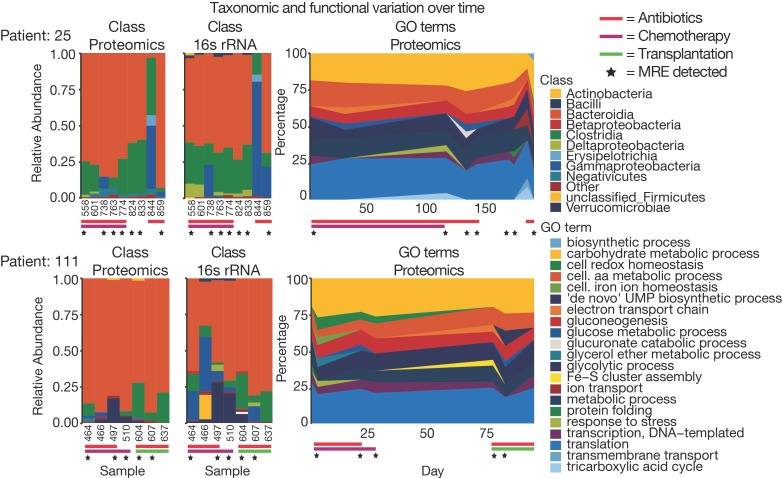
The microbiome has a strong impact on human health and disease and is, therefore, increasingly studied in a clinical context. Metaproteomics is also attracting considerable attention, and such data can be efficiently generated today owing to improvements in mass spectrometry-based proteomics. As we will discuss in this study, there are still major challenges notably in data analysis that need to be overcome. Here, we analyzed 212 fecal samples from 56 hospitalized acute leukemia patients with multidrug-resistant Enterobactericeae (MRE) gut colonization using metagenomics and metaproteomics. This is one of the largest clinical metaproteomic studies to date, and the first metaproteomic study addressing the gut microbiome in MRE colonized acute leukemia patients. Based on this substantial data set, we discuss major current limitations in clinical metaproteomic data analysis to provide guidance to researchers in the field. Notably, the results show that public metagenome databases are incomplete and that sample-specific metagenomes improve results. Furthermore, biological variation is tremendous which challenges clinical study designs and argues that longitudinal measurements of individual patients are a valuable future addition to the analysis of patient cohorts.
Keywords: clinical proteomics; data analysis; human gut microbiome; mass spectrometry; metaproteome; multi-omics; multidrug-resistant Enterobacteriaceae; proteomics.
Conflict of interest statement
B.K. and M.W. are founders and shareholders of OmicScouts. They have no operational role in the company. The company was not involved in this study. All other authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
