

Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis
- PMID: 30626590
- PMCID: PMC6643997
- DOI: 10.1158/2159-8290.CD-18-0567
Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis
Erratum in
-
Correction: Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis.Cancer Discov. 2024 Sep 4;14(9):1757. doi: 10.1158/2159-8290.CD-24-0865. Cancer Discov. 2024. PMID: 39228300 No abstract available.
Abstract
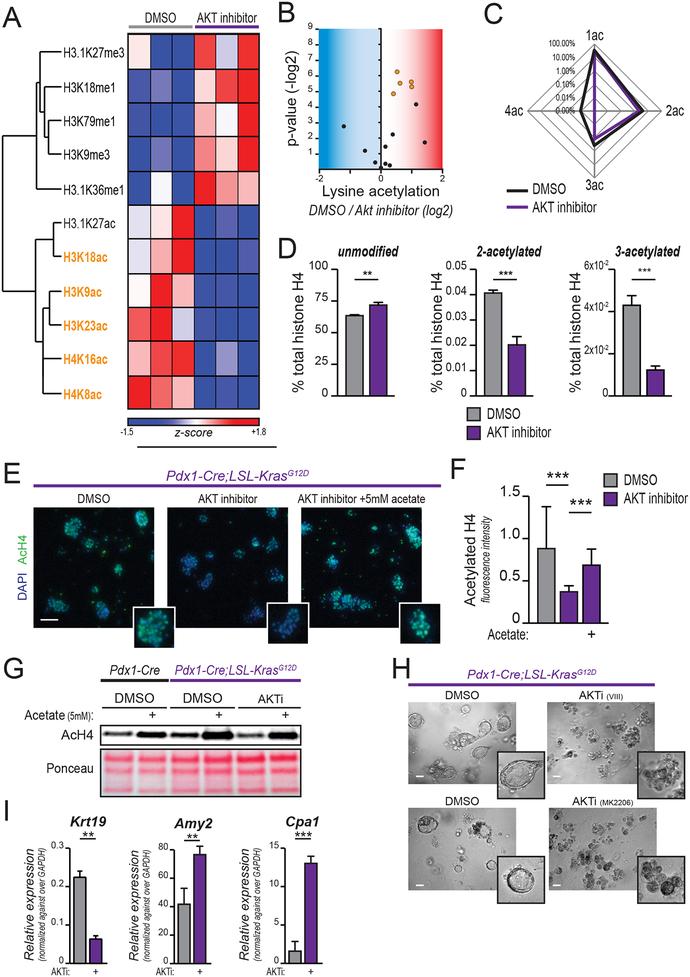
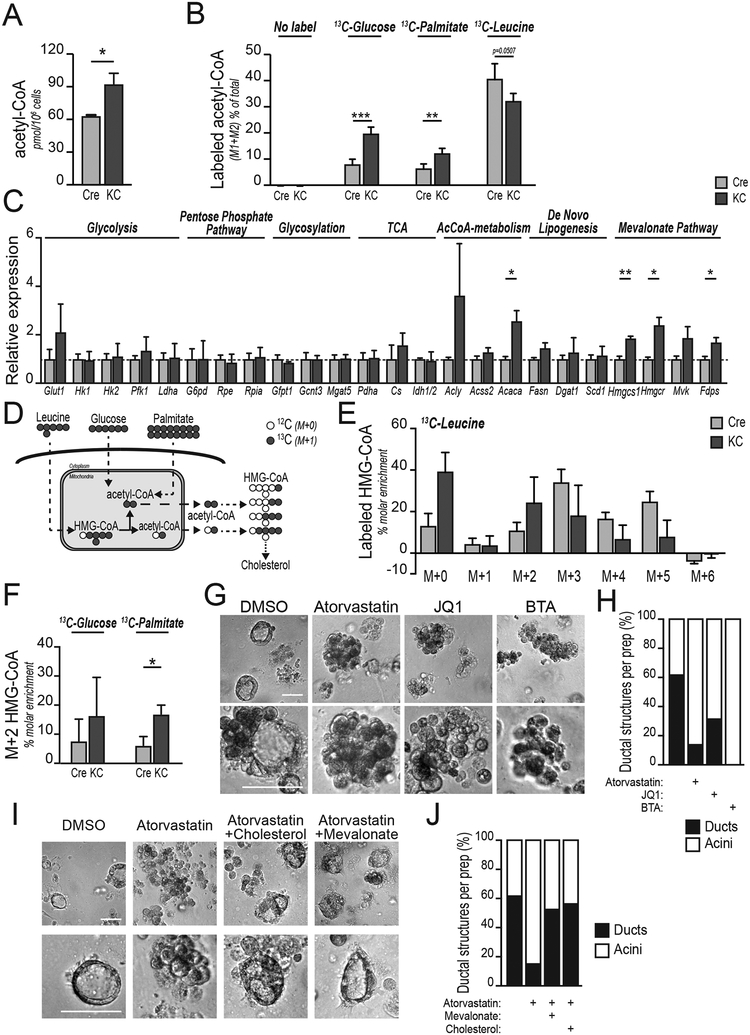
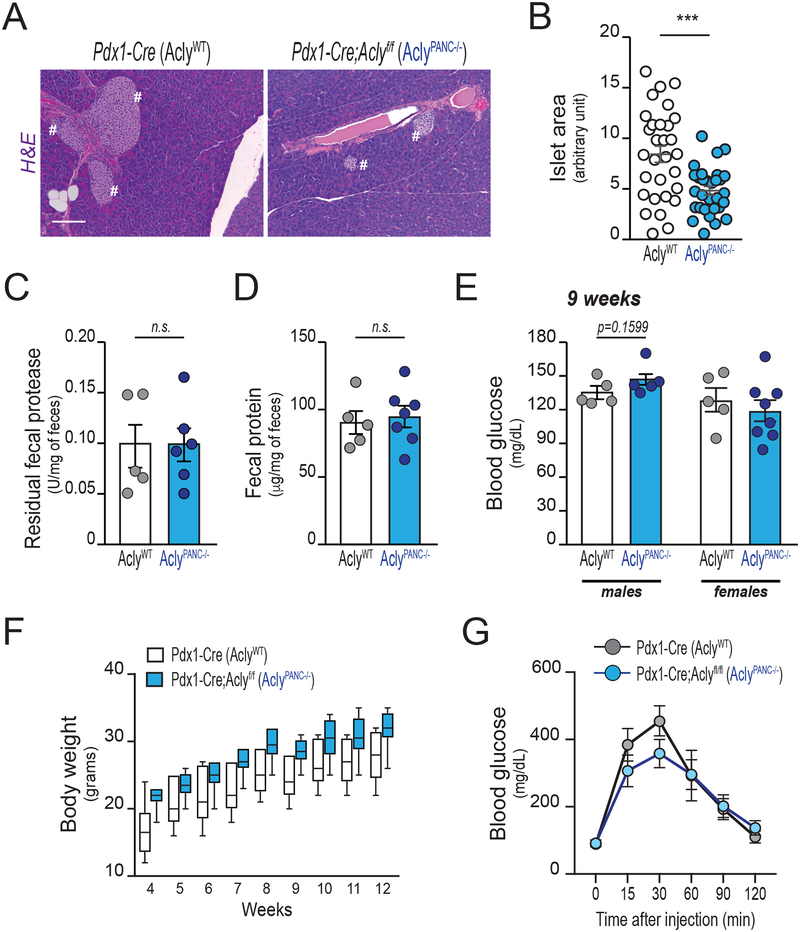
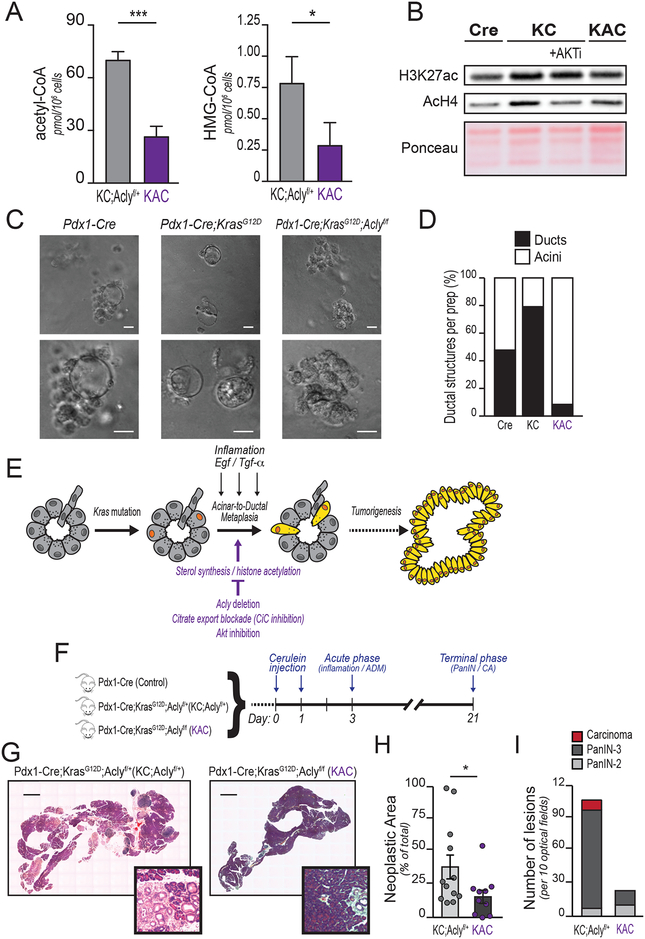
Pancreatic ductal adenocarcinoma (PDA) has a poor prognosis, and new strategies for prevention and treatment are urgently needed. We previously reported that histone H4 acetylation is elevated in pancreatic acinar cells harboring Kras mutations prior to the appearance of premalignant lesions. Because acetyl-CoA abundance regulates global histone acetylation, we hypothesized that altered acetyl-CoA metabolism might contribute to metabolic or epigenetic alterations that promote tumorigenesis. We found that acetyl-CoA abundance is elevated in KRAS-mutant acinar cells and that its use in the mevalonate pathway supports acinar-to-ductal metaplasia (ADM). Pancreas-specific loss of the acetyl-CoA-producing enzyme ATP-citrate lyase (ACLY) accordingly suppresses ADM and tumor formation. In PDA cells, growth factors promote AKT-ACLY signaling and histone acetylation, and both cell proliferation and tumor growth can be suppressed by concurrent BET inhibition and statin treatment. Thus, KRAS-driven metabolic alterations promote acinar cell plasticity and tumor development, and targeting acetyl-CoA-dependent processes exerts anticancer effects. SIGNIFICANCE: Pancreatic cancer is among the deadliest of human malignancies. We identify a key role for the metabolic enzyme ACLY, which produces acetyl-CoA, in pancreatic carcinogenesis. The data suggest that acetyl-CoA use for histone acetylation and in the mevalonate pathway facilitates cell plasticity and proliferation, suggesting potential to target these pathways.See related commentary by Halbrook et al., p. 326.This article is highlighted in the In This Issue feature, p. 305.
©2019 American Association for Cancer Research.
Conflict of interest statement
Figures
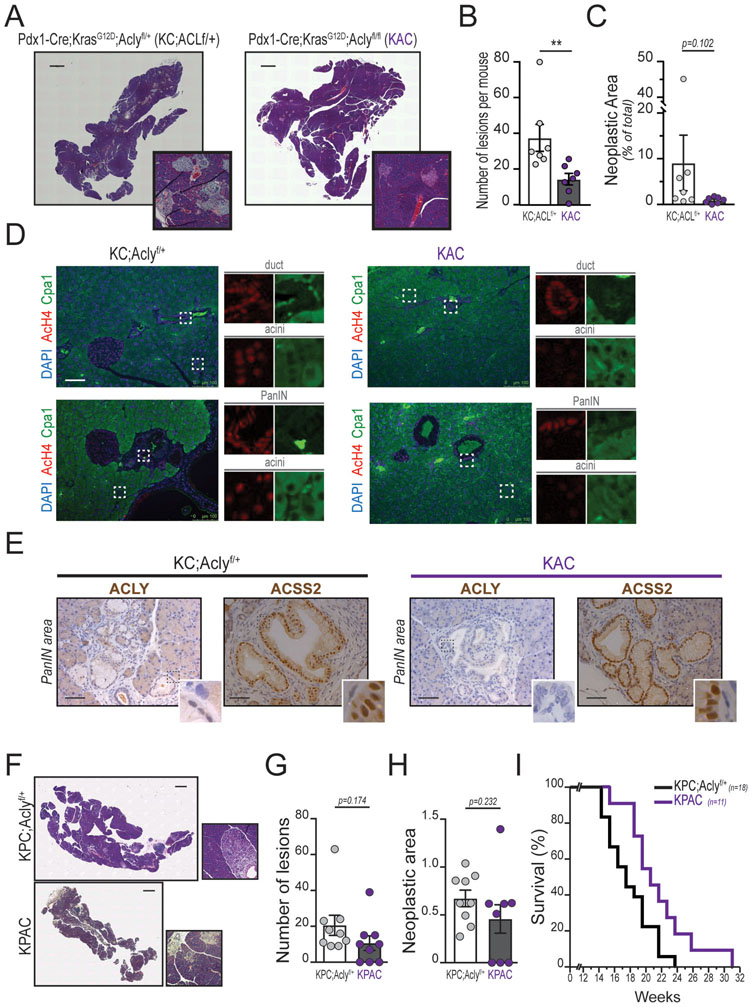
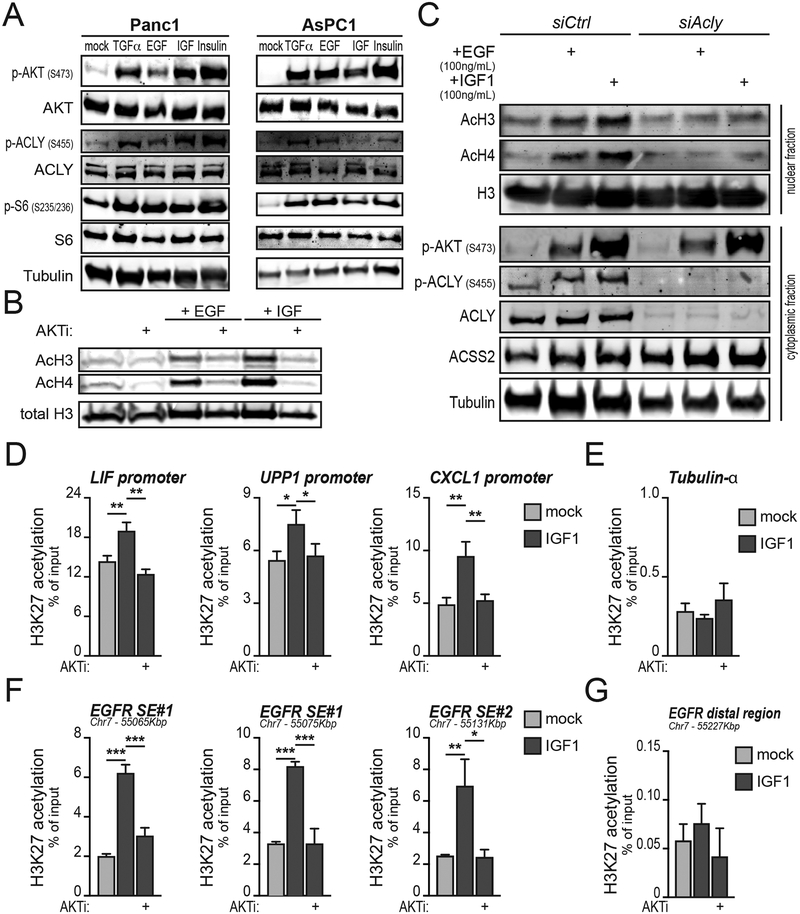
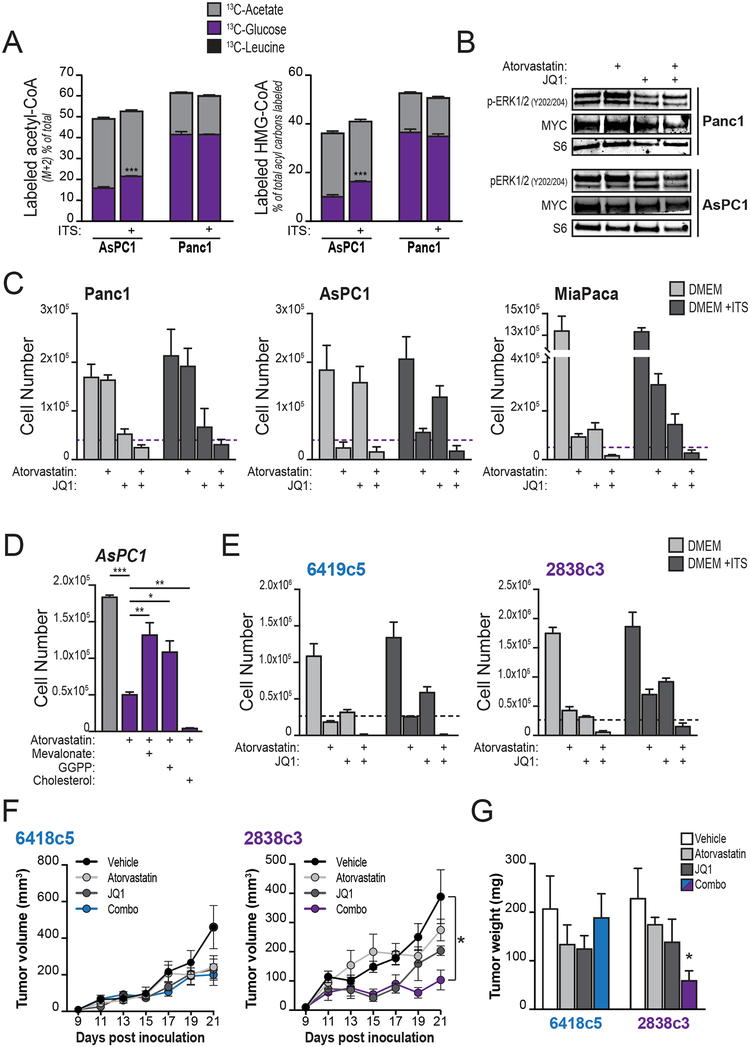
Comment in
-
Metabolism Drives Carcinogenesis and Maintenance of Pancreatic Tumors.Cancer Discov. 2019 Mar;9(3):326-328. doi: 10.1158/2159-8290.CD-19-0034. Cancer Discov. 2019. PMID: 30824487
References
-
- Borazanci E, Dang CV, Robey RW, Bates SE, Chabot JA, Von Hoff DD. Pancreatic Cancer: “A Riddle Wrapped in a Mystery inside an Enigma”. Clin Cancer Res. 2017;23:1629–1637. Available from: http://clincancerres.aacrjournals.org/content/23/7/1629.abstract - PubMed
-
- Noone AM, Howlander N, Krapcho M, Miller D, Brest A, et al. , SEER Cancer Statistics Review, 1975–2015, National Cancer Institute. Bethesda, MD: Available from: https://seer.cancer.gov/csr/1975_2015/, based on November 2017 SEER data submission, posted to the SEER web site, April 2018.
-
- Halbrook CJ, Lyssiotis CA. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell. Elsevier; 2017. page 5–19. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
