

Randomized trial of l-serine in patients with hereditary sensory and autonomic neuropathy type 1
- PMID: 30626650
- PMCID: PMC6345118
- DOI: 10.1212/WNL.0000000000006811
Randomized trial of l-serine in patients with hereditary sensory and autonomic neuropathy type 1
Abstract
Objective: To evaluate the safety and efficacy of l-serine in humans with hereditary sensory autonomic neuropathy type I (HSAN1).
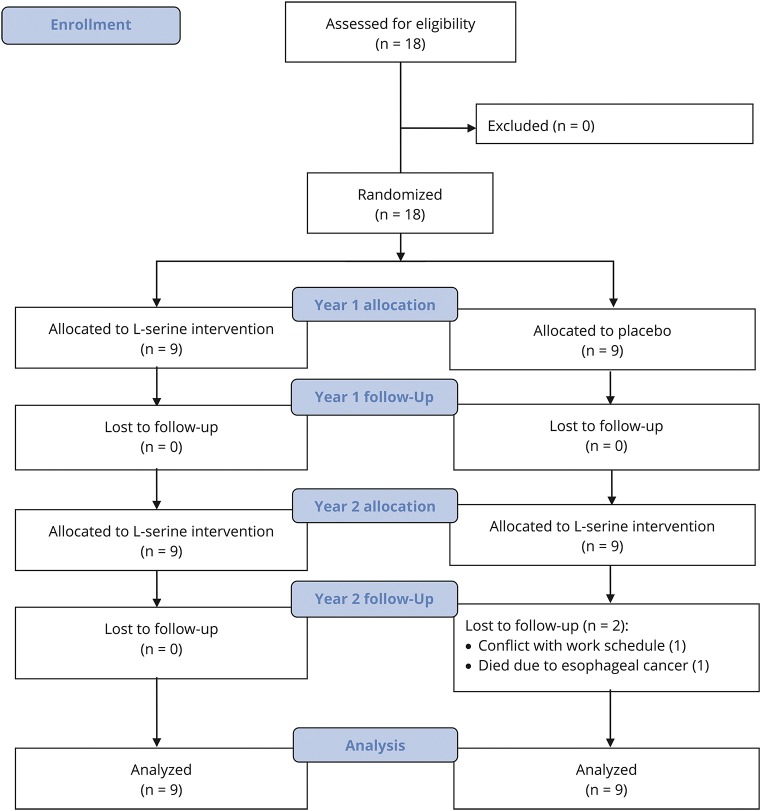
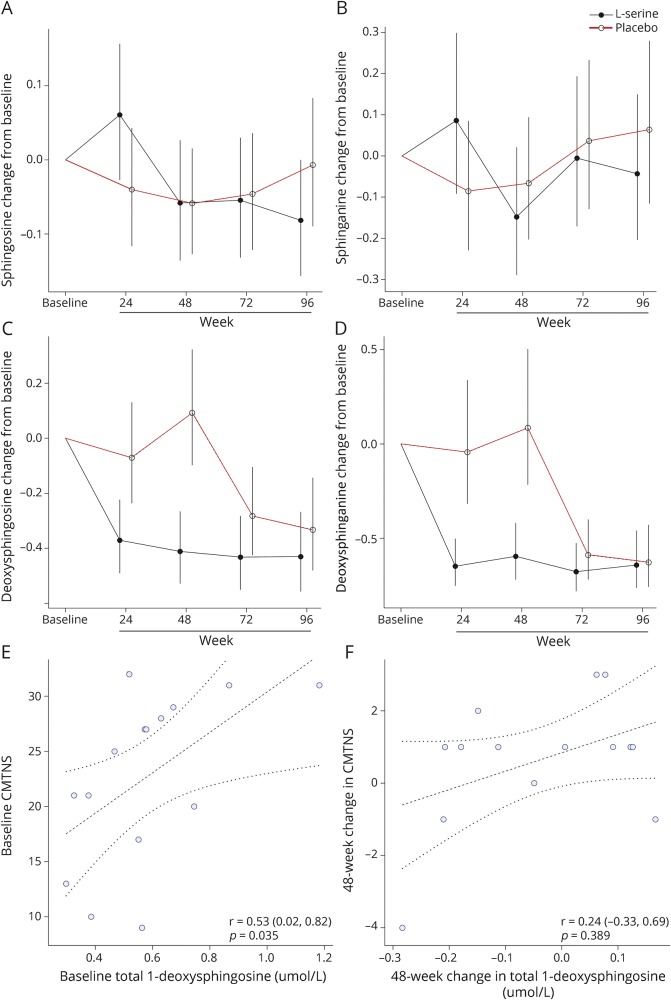
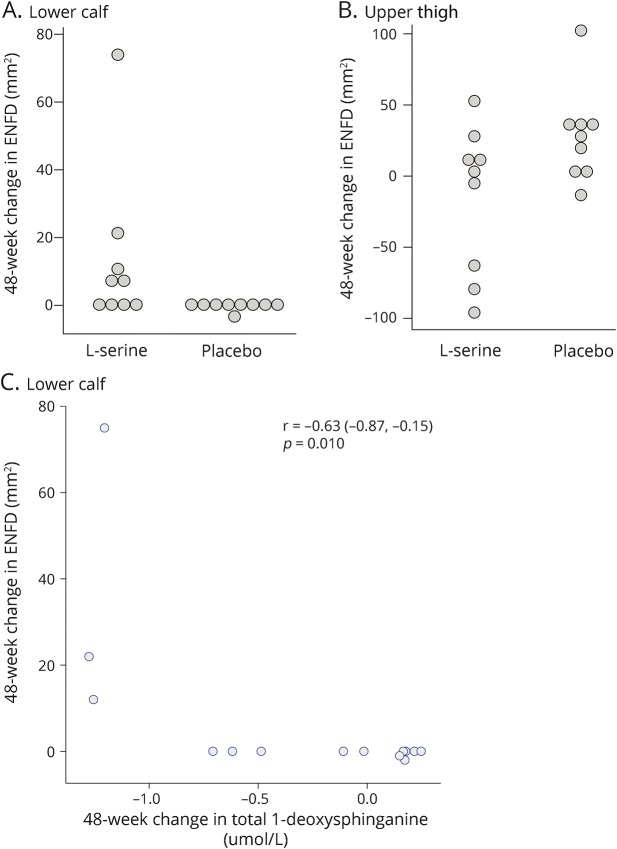
Methods: In this randomized, placebo-controlled, parallel-group trial with open-label extension, patients aged 18-70 years with symptomatic HSAN1 were randomized to l-serine (400 mg/kg/day) or placebo for 1 year. All participants received l-serine during the second year. The primary outcome measure was the Charcot-Marie-Tooth Neuropathy Score version 2 (CMTNS). Secondary outcomes included plasma sphingolipid levels, epidermal nerve fiber density, electrophysiologic measurements, patient-reported measures, and adverse events.
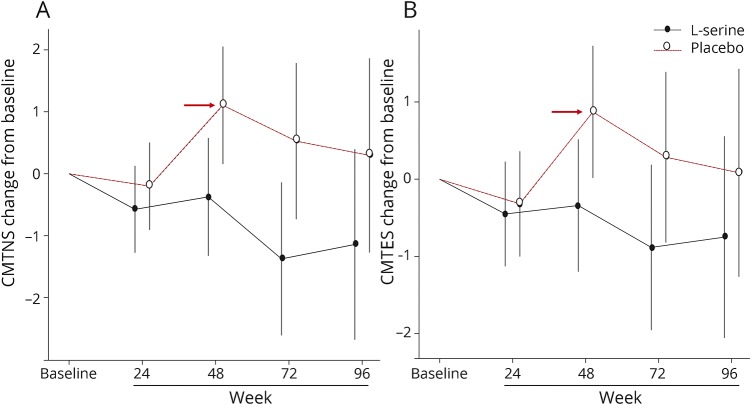
Results: Between August 2013 and April 2014, we enrolled and randomized 18 participants, 16 of whom completed the study. After 1 year, the l-serine group experienced improvement in CMTNS relative to the placebo group (-1.5 units, 95% CI -2.8 to -0.1, p = 0.03), with evidence of continued improvement in the second year of treatment (-0.77, 95% CI -1.67 to 0.13, p = 0.09). Concomitantly, deoxysphinganine levels dropped in l-serine-treated but not placebo-treated participants (59% decrease vs 11% increase; p < 0.001). There were no serious adverse effects related to l-serine.
Conclusion: High-dose oral l-serine supplementation appears safe in patients with HSAN1 and is potentially effective at slowing disease progression.
Clinicaltrialsgov identifier: NCT01733407.
Classification of evidence: This study provides Class I evidence that high-dose oral l-serine supplementation significantly slows disease progression in patients with HSAN1.
Copyright © 2019 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Figures
References
-
- Houlden H, King R, Blake J, et al. Clinical, pathological and genetic characterization of hereditary sensory and autonomic neuropathy type 1 (HSAN I). Brain 2006;129:411–425. - PubMed
-
- Bejaoui K, Wu C, Scheffler MD, et al. SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat Genet 2001;27:261–262. - PubMed
-
- Dawkins JL, Hulme DJ, Brahmbhatt SB, Auer-Grumbach M, Nicholson GA. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet 2001;27:309–312. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous