

BACE2, a conditional β-secretase, contributes to Alzheimer's disease pathogenesis
- PMID: 30626751
- PMCID: PMC6485359
- DOI: 10.1172/jci.insight.123431
BACE2, a conditional β-secretase, contributes to Alzheimer's disease pathogenesis
Abstract
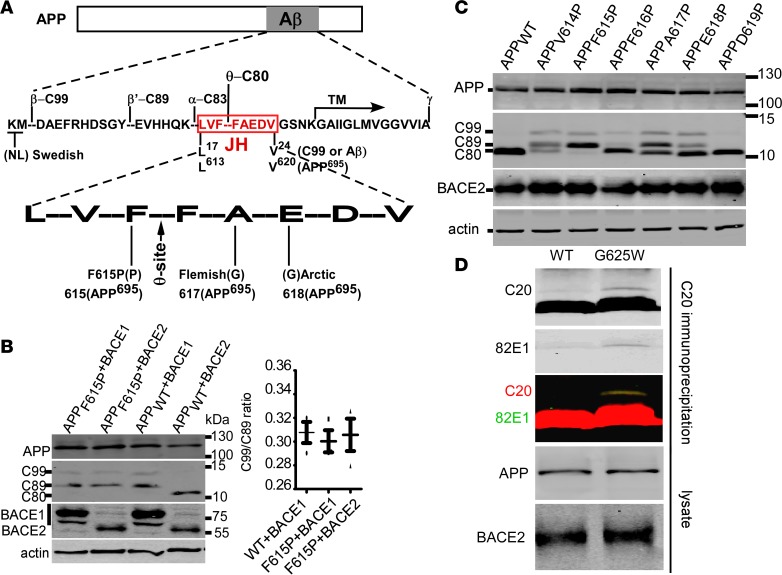
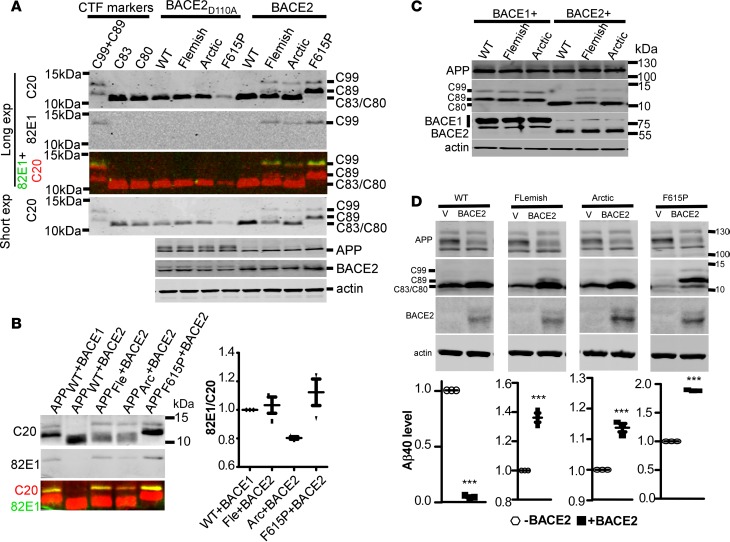
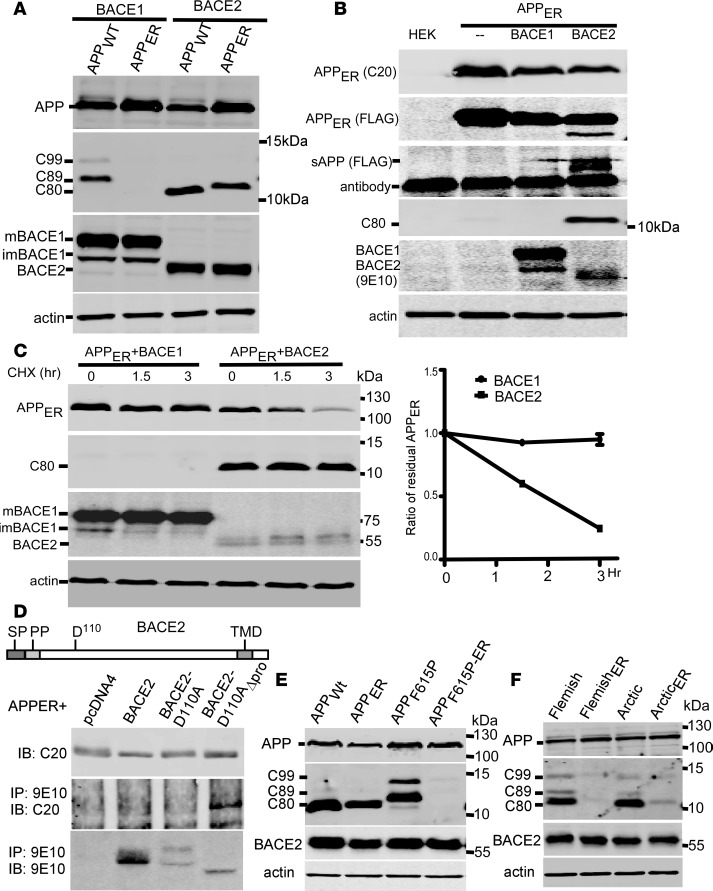
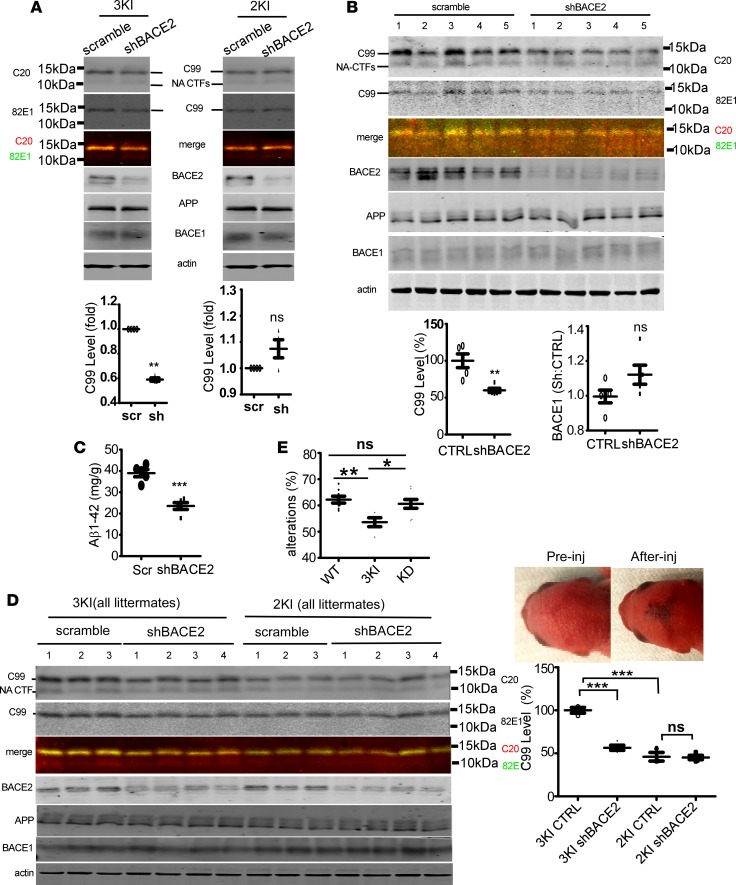
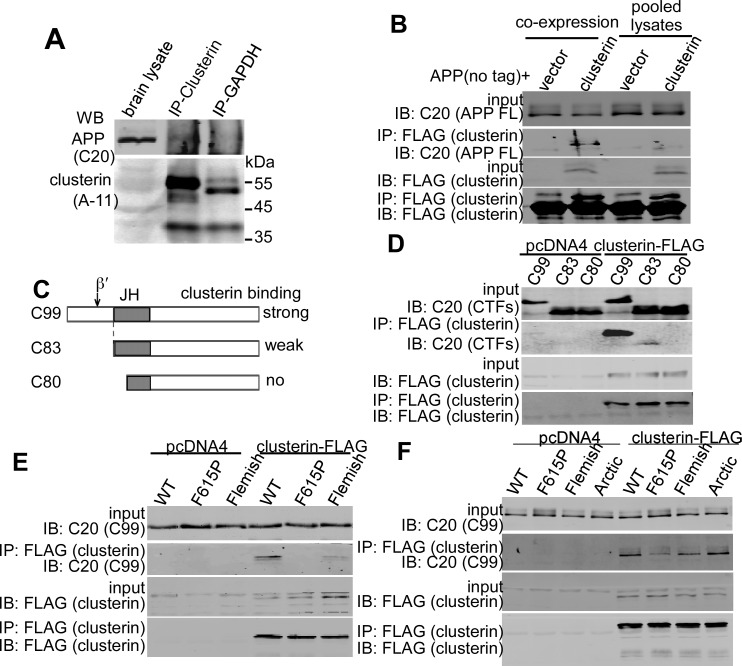
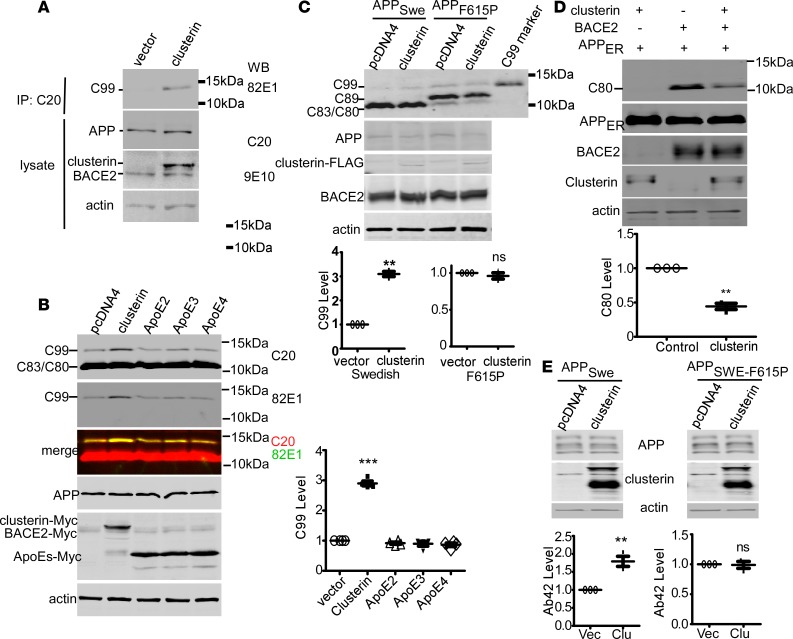
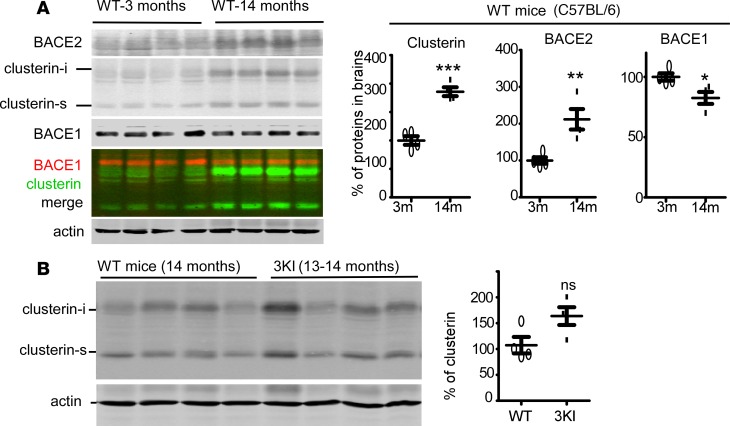
Deposition of amyloid-β protein (Aβ) to form neuritic plaques is the characteristic neuropathology of Alzheimer's disease (AD). Aβ is generated from amyloid precursor protein (APP) by β- and γ-secretase cleavages. BACE1 is the β-secretase and its inhibition induces severe side effects, whereas its homolog BACE2 normally suppresses Aβ by cleaving APP/Aβ at the θ-site (Phe20) within the Aβ domain. Here, we report that BACE2 also processes APP at the β site, and the juxtamembrane helix (JH) of APP inhibits its β-secretase activity, enabling BACE2 to cleave nascent APP and aggravate AD symptoms. JH-disrupting mutations and clusterin binding to JH triggered BACE2-mediated β-cleavage. Both BACE2 and clusterin were elevated in aged mouse brains, and enhanced β-cleavage during aging. Therefore, BACE2 contributes to AD pathogenesis as a conditional β-secretase and could be a preventive and therapeutic target for AD without the side effects of BACE1 inhibition.
Keywords: Alzheimer’s disease; Neuroscience.
Conflict of interest statement
Figures
References
-
- Kang Y, et al. Nutritional deficiency in early life facilitates aging-associated cognitive decline. Curr Alzheimer Res. 2017;14(8):841–849. - PubMed
-
- Zhang S, et al. A presenilin-1 mutation causes Alzheimer disease without affecting Notch signaling [published online ahead of print]. Mol Psychiatry. doi: 10.1038/s41380-018-0101-x. - PubMed
LinkOut - more resources
Full Text Sources
