

Multi-step genomic dissection of a suspected intra-hospital Helicobacter cinaedi outbreak
- PMID: 30629483
- PMCID: PMC6412056
- DOI: 10.1099/mgen.0.000236
Multi-step genomic dissection of a suspected intra-hospital Helicobacter cinaedi outbreak
Abstract
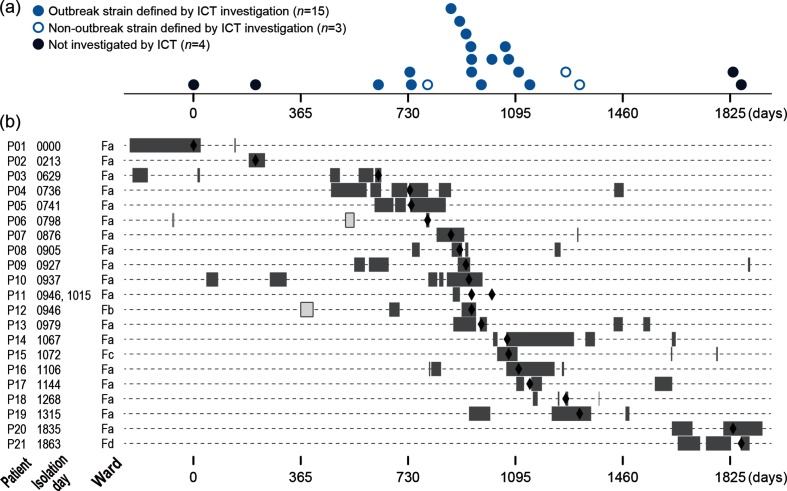
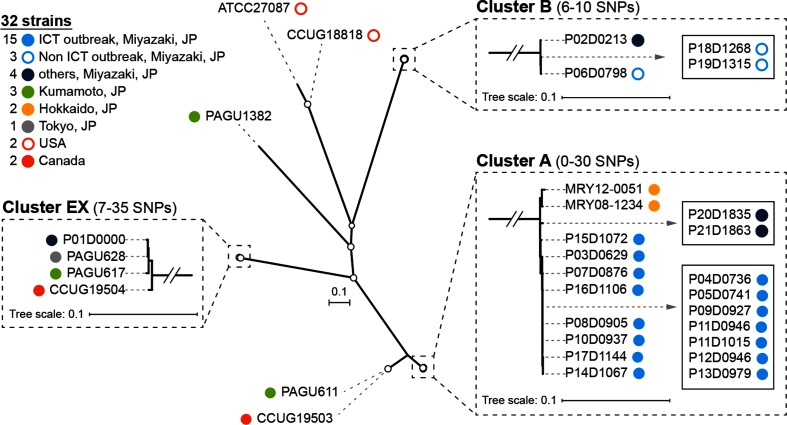
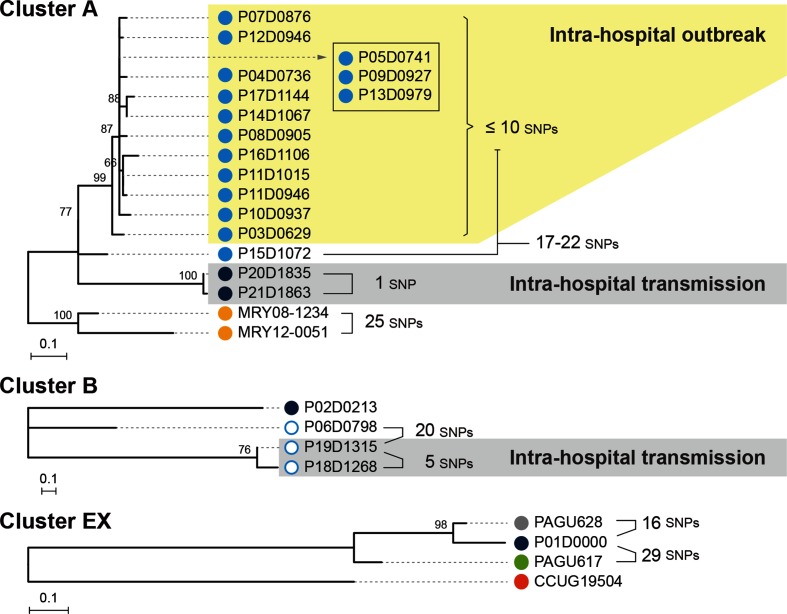
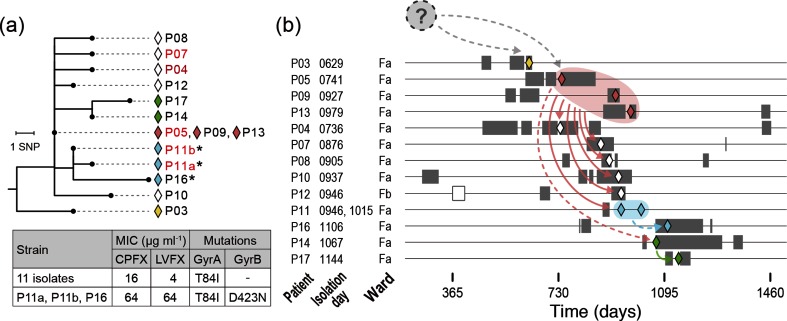
Helicobacter cinaedi is an emerging pathogen causing bacteraemia and cellulitis. Nosocomial transmission of this microbe has been described, but detailed molecular-epidemiological analyses have not been performed. Here, we describe the results of a multi-step genome-wide phylogenetic analysis of a suspected intra-hospital outbreak of H. cinaedi that occurred in a hospital in Japan. The outbreak was recognized by the infectious control team (ICT) of the hospital as a sudden increase in H. cinaedi bacteraemia. ICT defined this outbreak case based on 16S rRNA sequence data and epidemiological information, but were unable to determine the source and route of the infections. We therefore re-investigated this case using whole-genome sequencing (WGS). We first performed a species-wide analysis using publicly available genome sequences to understand the level of genomic diversity of this under-studied species. The clusters identified were then separately analysed using the genome sequence of a representative strain in each cluster as a reference. These analyses provided a high-level phylogenetic resolution of each cluster, identified a confident set of outbreak isolates, and discriminated them from other closely related but distinct clones, which were locally circulating and invaded the hospital during the same period. By considering the epidemiological data, possible strain transmission chains were inferred, which highlighted the role of asymptomatic carriers or environmental contamination. The emergence of a subclone with increased resistance to fluoroquinolones in the outbreak was also recognized. Our results demonstrate the impact of the use of a closely related genome as a reference to maximize the power of WGS.
Keywords: Helicobacter cinaedi; antimicrobial resistance; asymptomatic carrier; intra-hospital outbreak; multi-step phylogenetic analysis.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical