

Accurate prediction of cell type-specific transcription factor binding
- PMID: 30630522
- PMCID: PMC6327544
- DOI: 10.1186/s13059-018-1614-y
Accurate prediction of cell type-specific transcription factor binding
Abstract
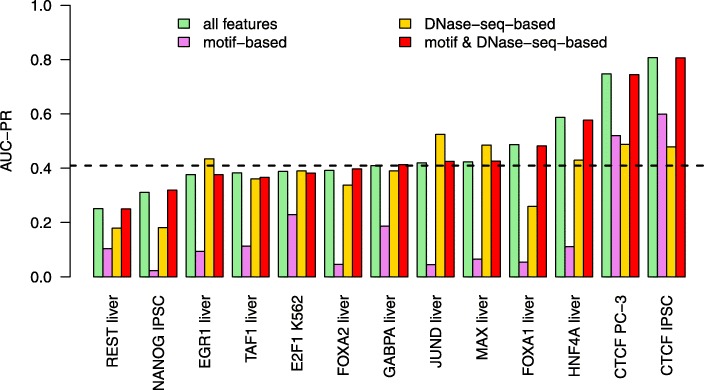
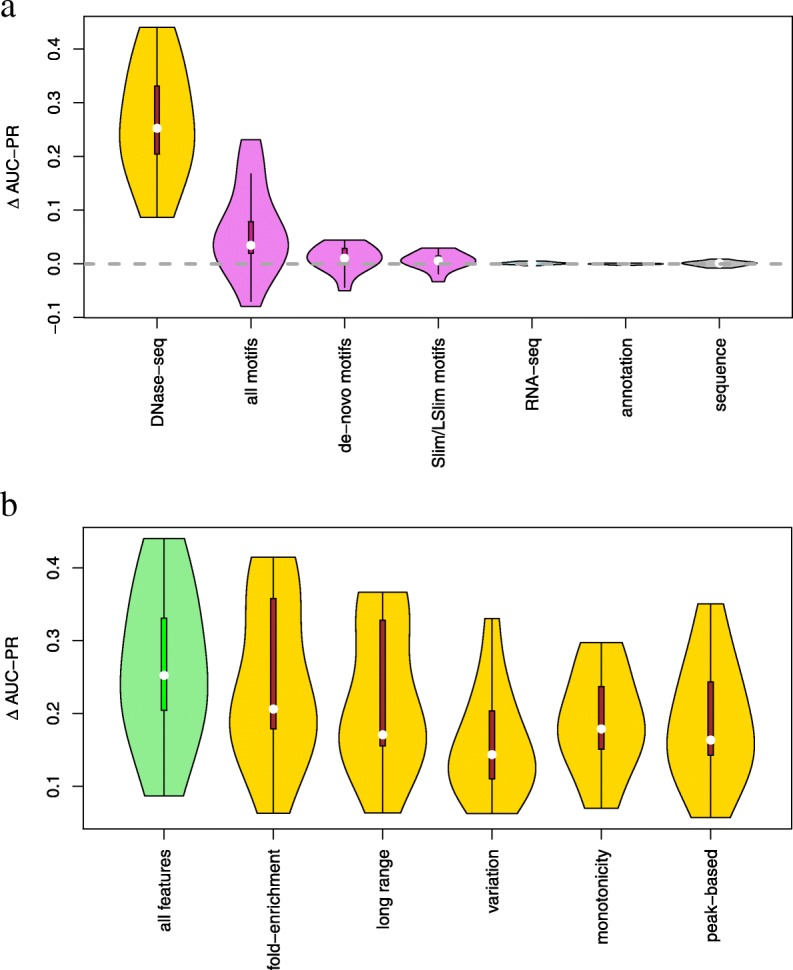
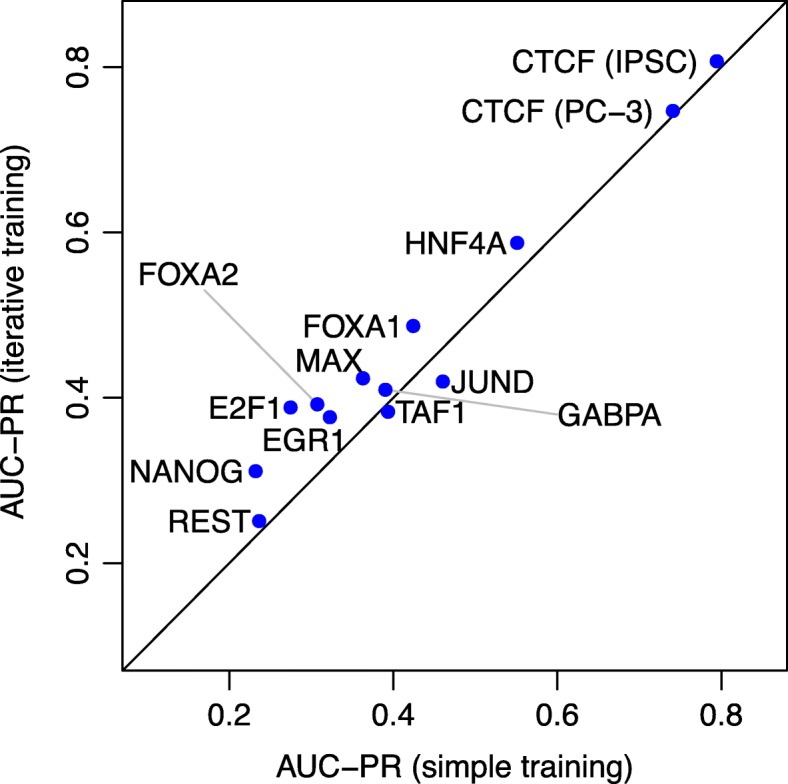
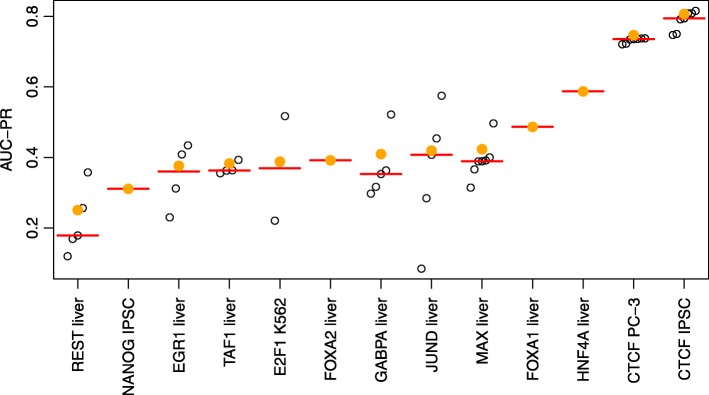
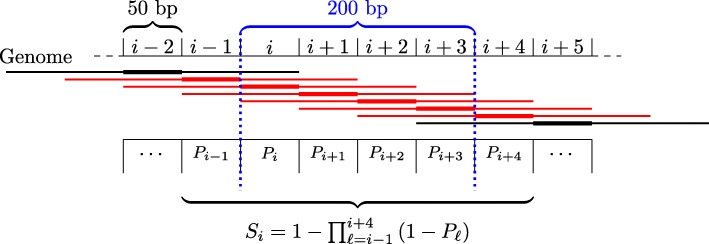
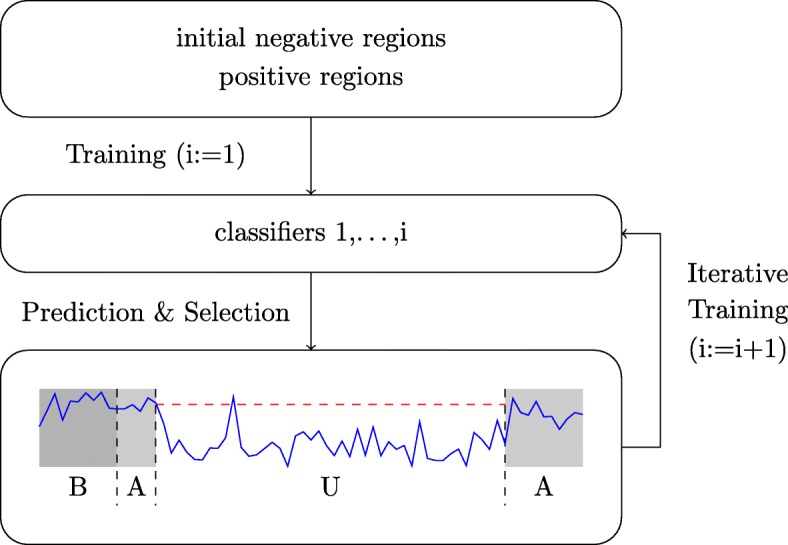
Prediction of cell type-specific, in vivo transcription factor binding sites is one of the central challenges in regulatory genomics. Here, we present our approach that earned a shared first rank in the "ENCODE-DREAM in vivo Transcription Factor Binding Site Prediction Challenge" in 2017. In post-challenge analyses, we benchmark the influence of different feature sets and find that chromatin accessibility and binding motifs are sufficient to yield state-of-the-art performance. Finally, we provide 682 lists of predicted peaks for a total of 31 transcription factors in 22 primary cell types and tissues and a user-friendly version of our approach, Catchitt, for download.
Keywords: Cell type-specific; ChIP-seq; DNase-seq; Machine learning; Transcription factors.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Berg OG, von Hippel PH. Selection of DNA binding sites by regulatory proteins: statistical-mechanical theory and application to operators and promoters. J Mol Biol. 1987;193(4):723–43. - PubMed
-
- Stormo GD, Fields DS. Specificity, free energy and information content in protein–DNA interactions. Trends Biochem Sci. 1998;23(3):109–13. - PubMed
-
- Wu J, Smith LT, Plass C, Huang TH-M. ChIP-chip comes of age for genome-wide functional analysis. Cancer Res. 2006;66(14):6899–902. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
