

CRISPR-Cas9 Circular Permutants as Programmable Scaffolds for Genome Modification
- PMID: 30633905
- PMCID: PMC6414052
- DOI: 10.1016/j.cell.2018.11.052
CRISPR-Cas9 Circular Permutants as Programmable Scaffolds for Genome Modification
Abstract
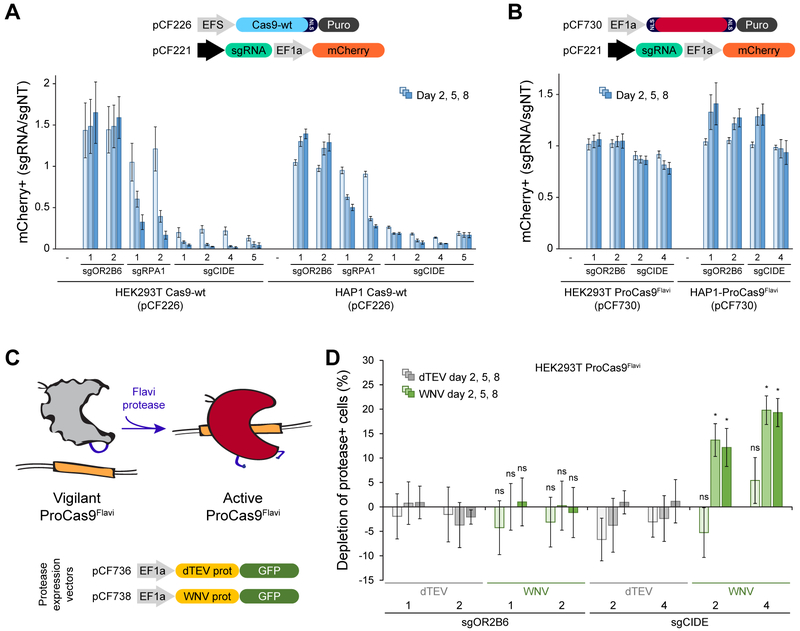
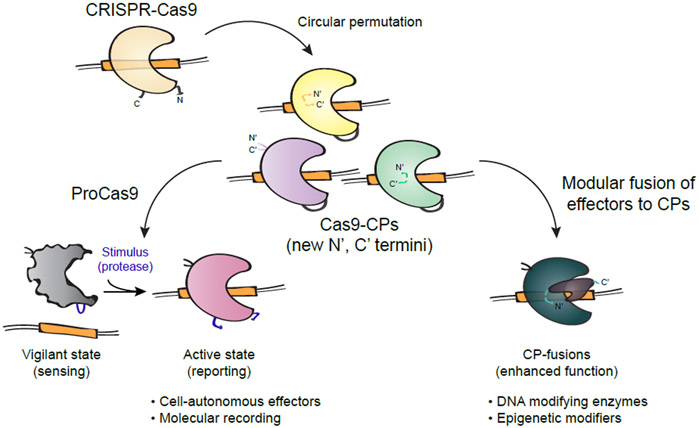
The ability to engineer natural proteins is pivotal to a future, pragmatic biology. CRISPR proteins have revolutionized genome modification, yet the CRISPR-Cas9 scaffold is not ideal for fusions or activation by cellular triggers. Here, we show that a topological rearrangement of Cas9 using circular permutation provides an advanced platform for RNA-guided genome modification and protection. Through systematic interrogation, we find that protein termini can be positioned adjacent to bound DNA, offering a straightforward mechanism for strategically fusing functional domains. Additionally, circular permutation enabled protease-sensing Cas9s (ProCas9s), a unique class of single-molecule effectors possessing programmable inputs and outputs. ProCas9s can sense a wide range of proteases, and we demonstrate that ProCas9 can orchestrate a cellular response to pathogen-associated protease activity. Together, these results provide a toolkit of safer and more efficient genome-modifying enzymes and molecular recorders for the advancement of precision genome engineering in research, agriculture, and biomedicine.
Keywords: CRISPR-Cas; Cas9-CP; ProCas9; circular permutation; fusion proteins; genome editing; protein engineering.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
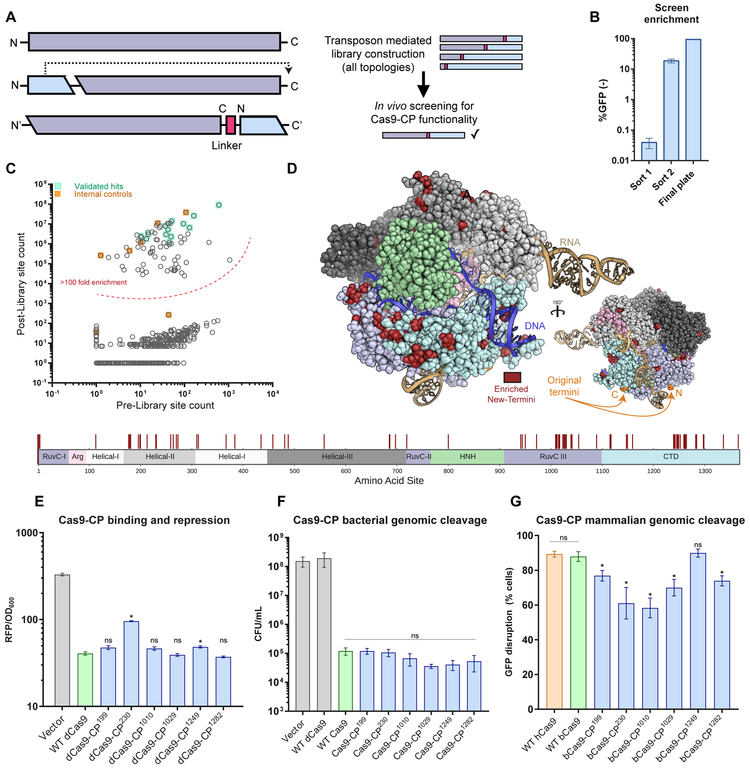
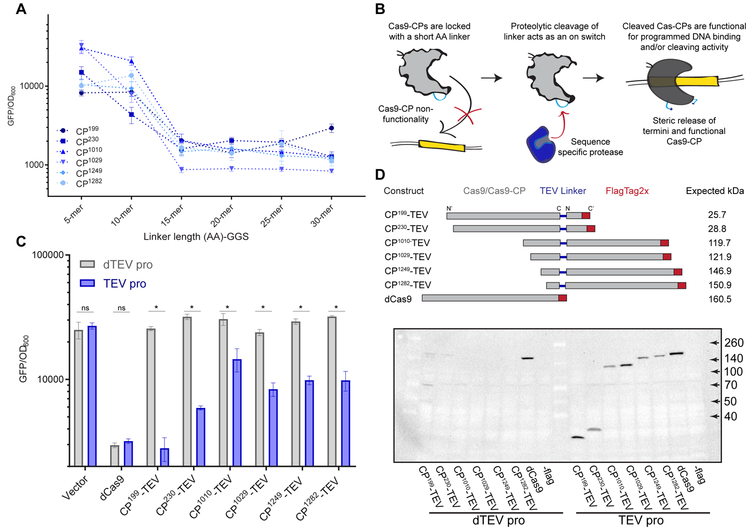
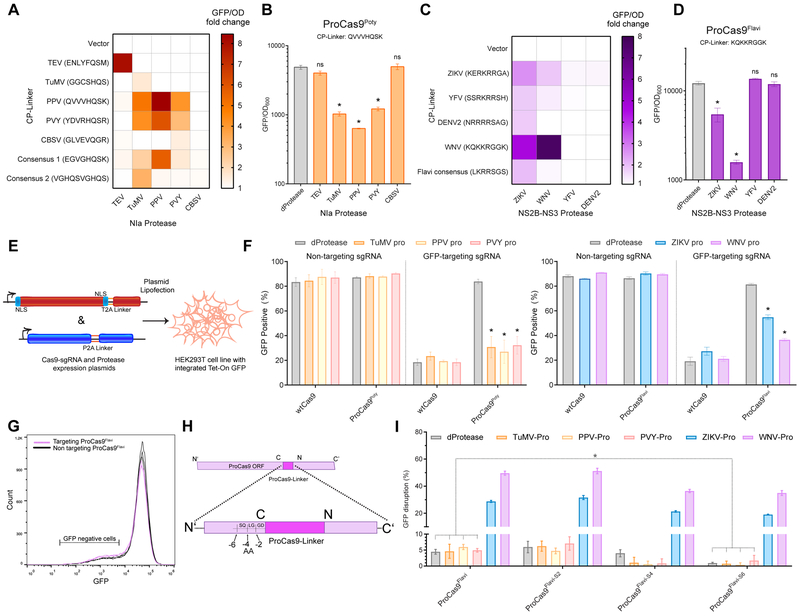
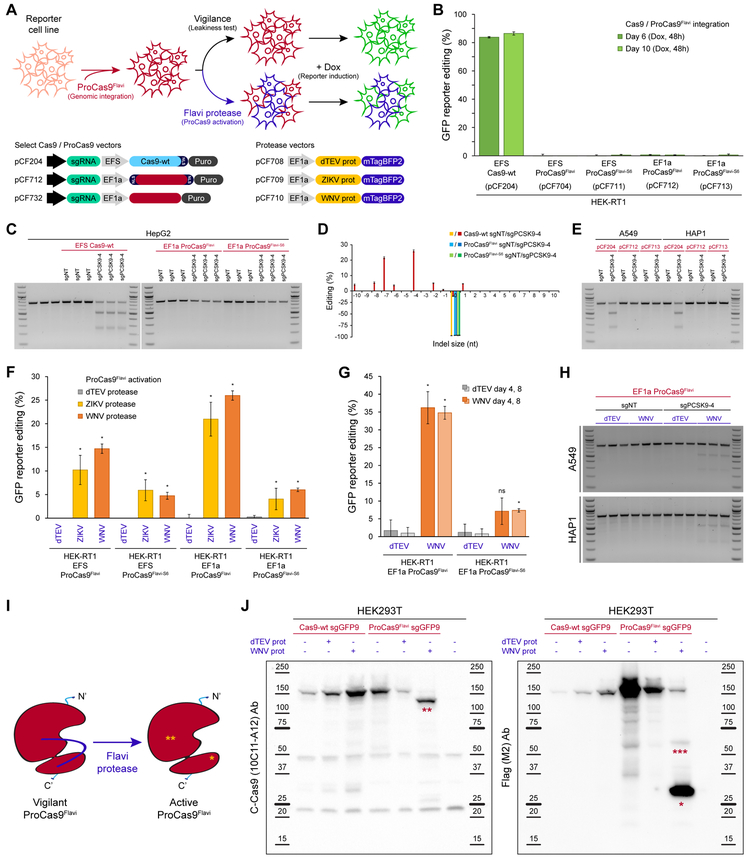
References
-
- Alfano JR, and Collmer A (2004). Type III secretion system effector proteins: double agents in bacterial disease and plant defense. Annu. Rev. Phytopathol 42, 385–414. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
