

Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding
- PMID: 30633912
- PMCID: PMC6363966
- DOI: 10.1016/j.cell.2018.11.046
Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding
Abstract
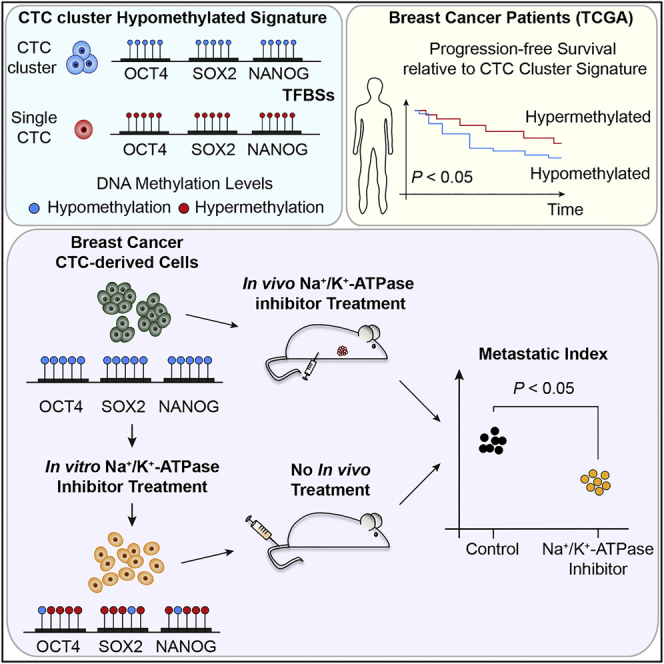
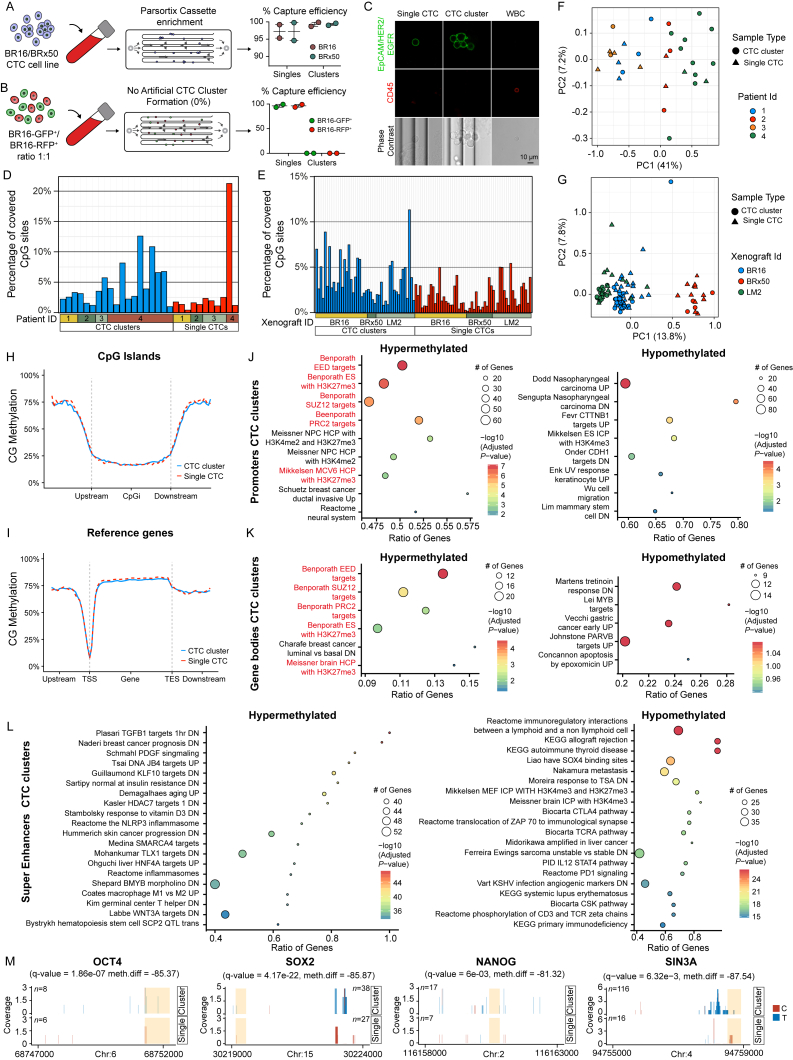
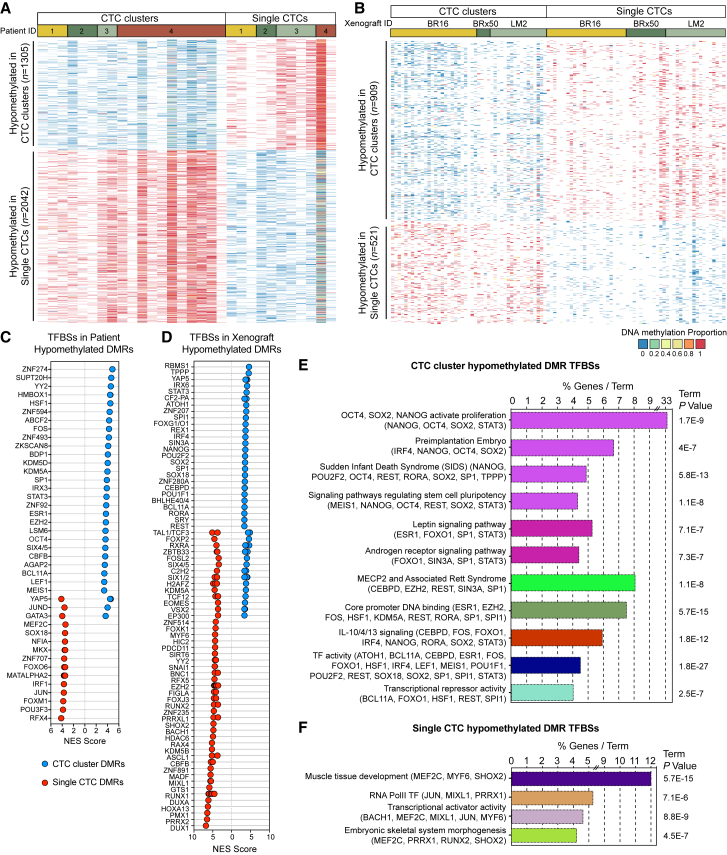
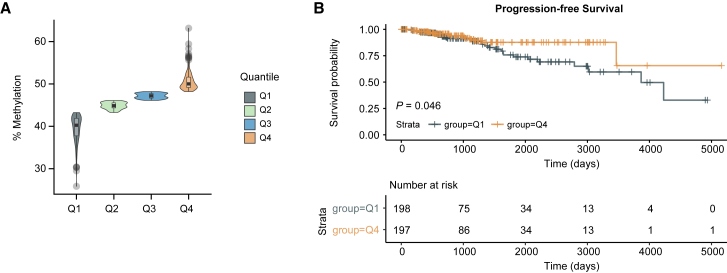
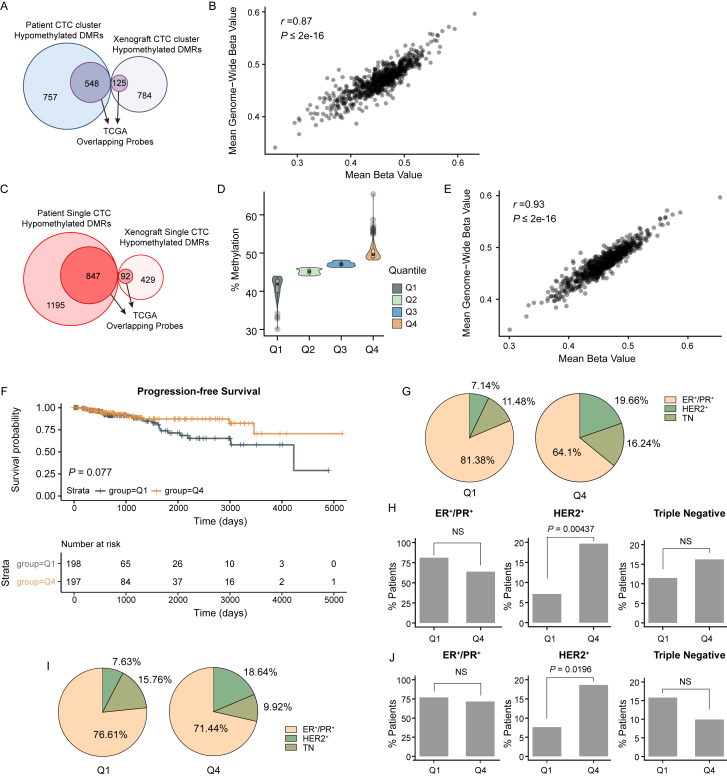
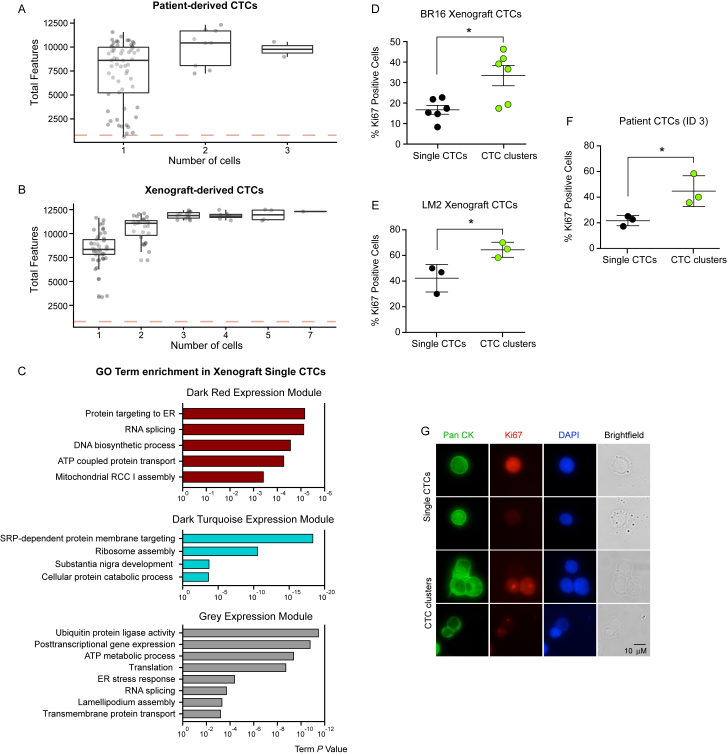
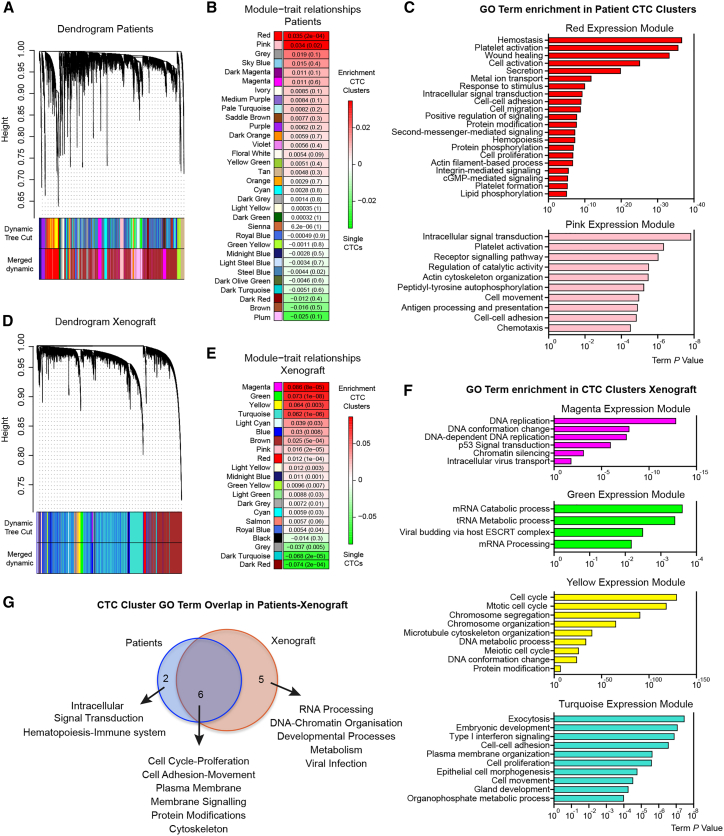
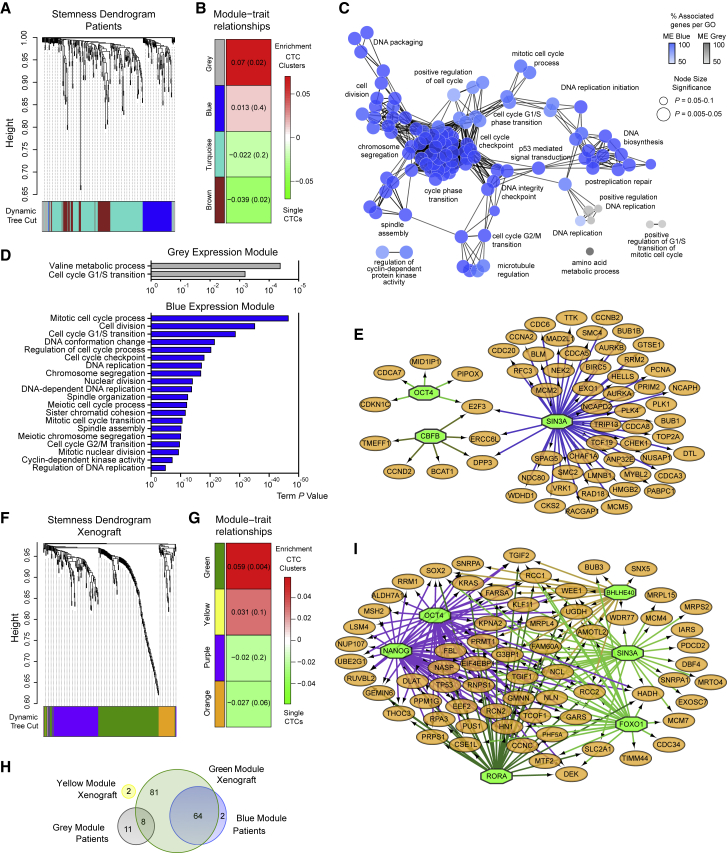
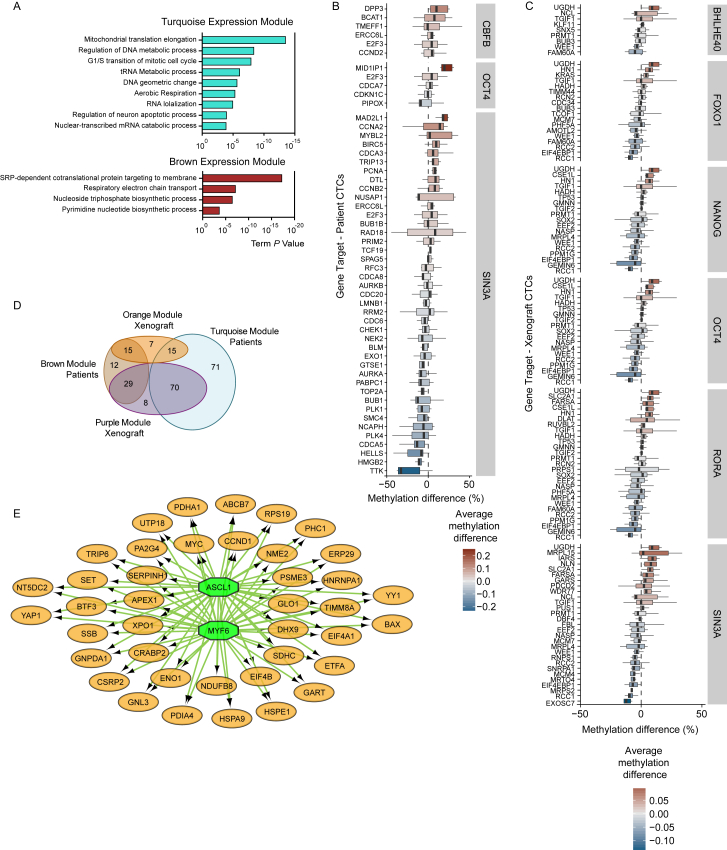
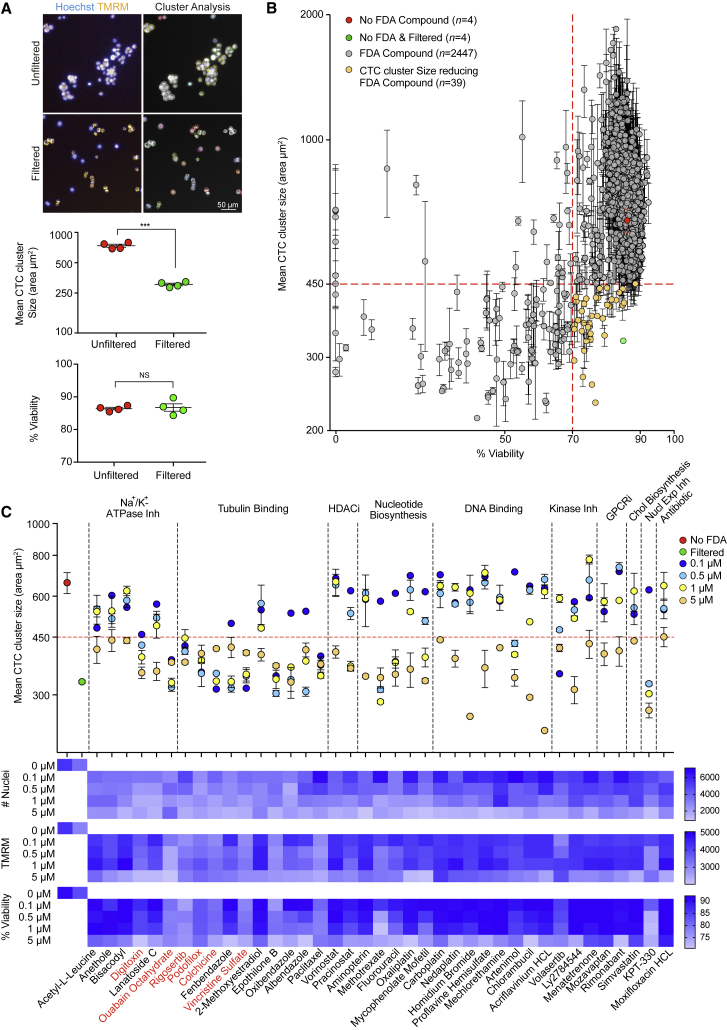
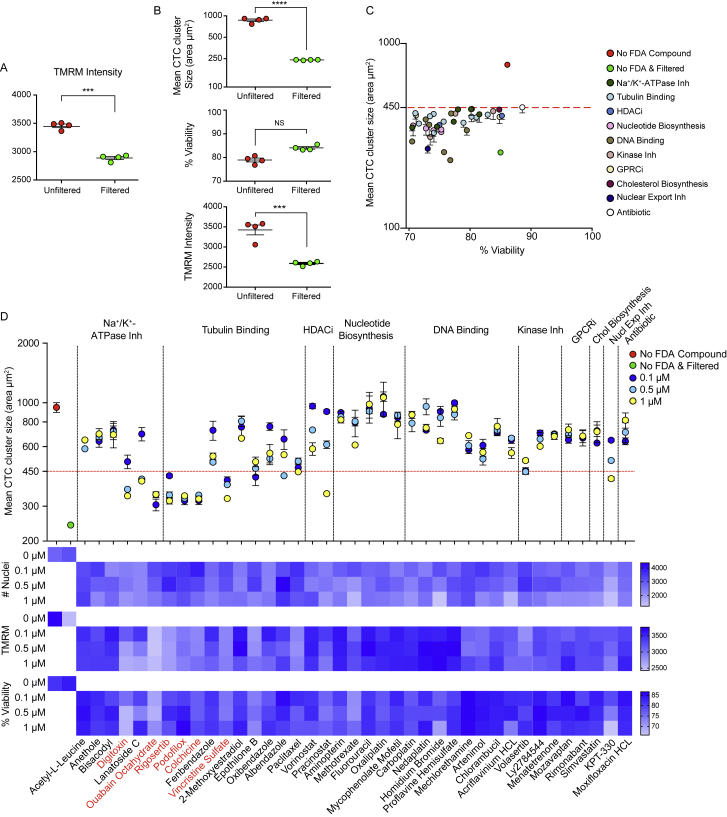
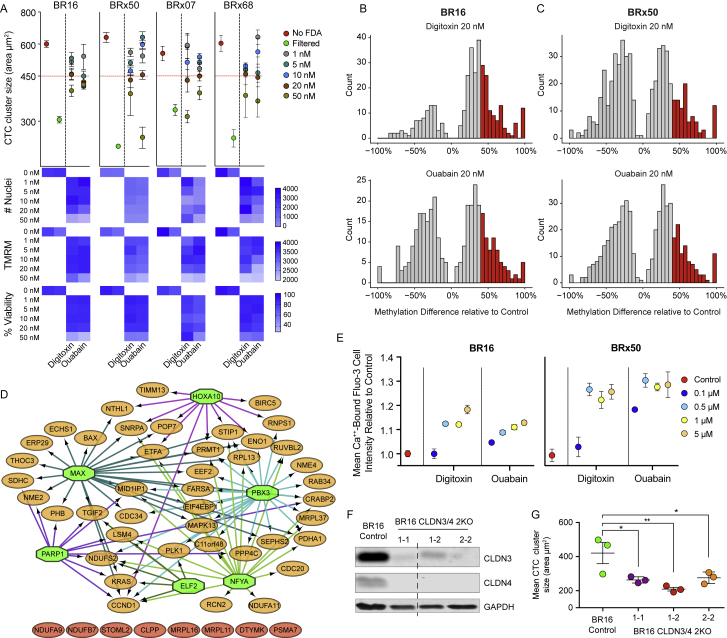
The ability of circulating tumor cells (CTCs) to form clusters has been linked to increased metastatic potential. Yet biological features and vulnerabilities of CTC clusters remain largely unknown. Here, we profile the DNA methylation landscape of single CTCs and CTC clusters from breast cancer patients and mouse models on a genome-wide scale. We find that binding sites for stemness- and proliferation-associated transcription factors are specifically hypomethylated in CTC clusters, including binding sites for OCT4, NANOG, SOX2, and SIN3A, paralleling embryonic stem cell biology. Among 2,486 FDA-approved compounds, we identify Na+/K+ ATPase inhibitors that enable the dissociation of CTC clusters into single cells, leading to DNA methylation remodeling at critical sites and metastasis suppression. Thus, our results link CTC clustering to specific changes in DNA methylation that promote stemness and metastasis and point to cluster-targeting compounds to suppress the spread of cancer.
Keywords: RNA sequencing; bisulfite sequencing; circulating tumor cell clusters; circulating tumor cells; drug screen; proliferation-associated transcription factors; single cell sequencing; stemness-associated transcription factors.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
References
-
- Aceto N., Toner M., Maheswaran S., Haber D.A. En route to metastasis: circulating tumor cell clusters and epithelial-to-mesenchymal transition. Trends Cancer. 2015;1:44–52. - PubMed
-
- Akalin A., Franke V., Vlahoviček K., Mason C.E., Schübeler D. Genomation: a toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics. 2015;31:1127–1129. - PubMed
-
- Alix-Panabières C., Pantel K. Circulating tumor cells: liquid biopsy of cancer. Clin. Chem. 2013;59:110–118. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
