

Phenotype prediction of Mohr-Tranebjaerg syndrome (MTS) by genetic analysis and initial auditory neuropathy
- PMID: 30634948
- PMCID: PMC6330410
- DOI: 10.1186/s12881-018-0741-3
Phenotype prediction of Mohr-Tranebjaerg syndrome (MTS) by genetic analysis and initial auditory neuropathy
Abstract
Background: Mohr-Tranebjaerg syndrome (MTS) is a rare X-linked recessive neurodegenerative disorder resulting in early-onset hearing impairment, gradual dystonia and optic atrophy. MTS is caused by variations in the nuclear TIMM8A gene, which is involved in mitochondrial transport of metabolites. This study aimed to identify the pathogenic gene variations in three Chinese families associated with predicted MTS with or without X-linked agammaglobulinaemia.
Methods: Otologic examinations, vestibular, neurological, optical and other clinical evaluations were conducted on the family members. Targeted genes capture combining next generation sequencing (NGS) was performed, and then Sanger sequencing was used to confirm the causative variation.
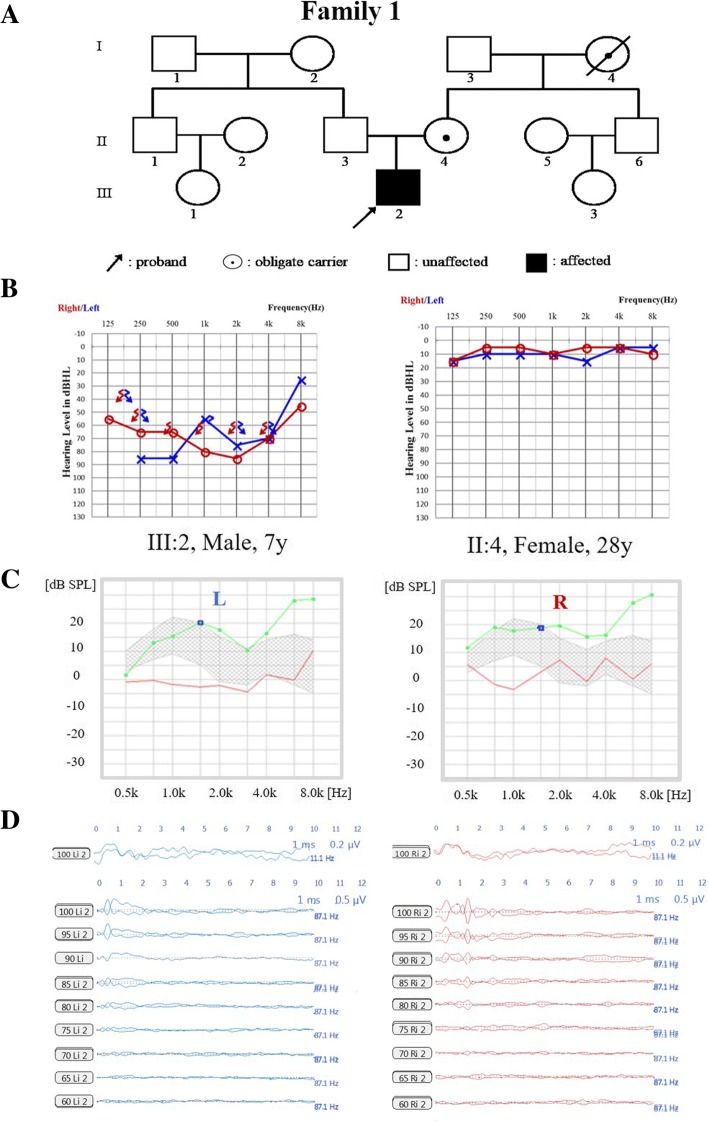
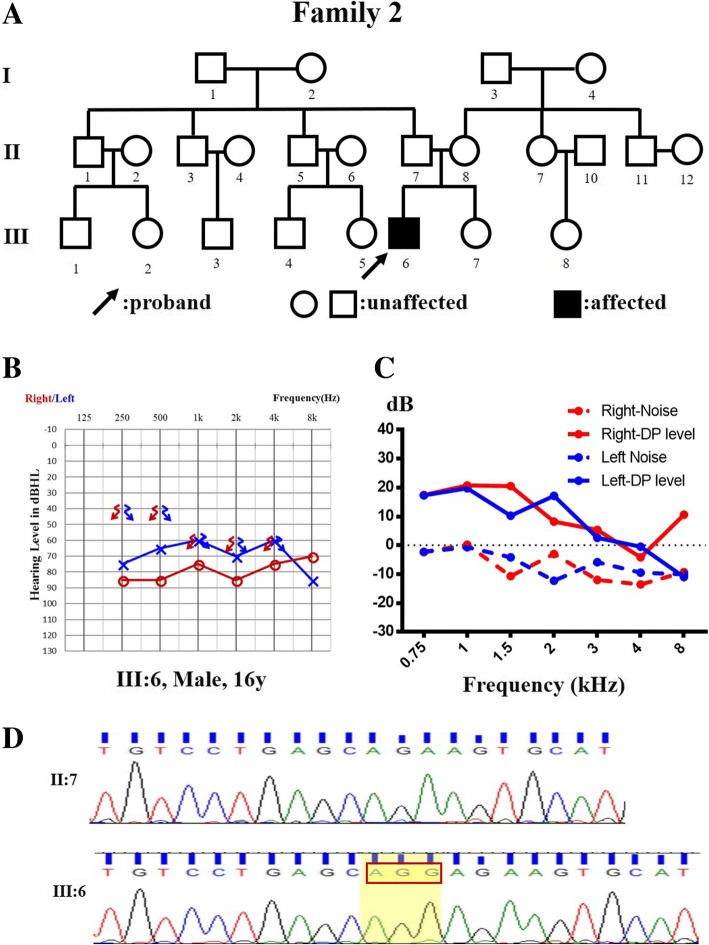
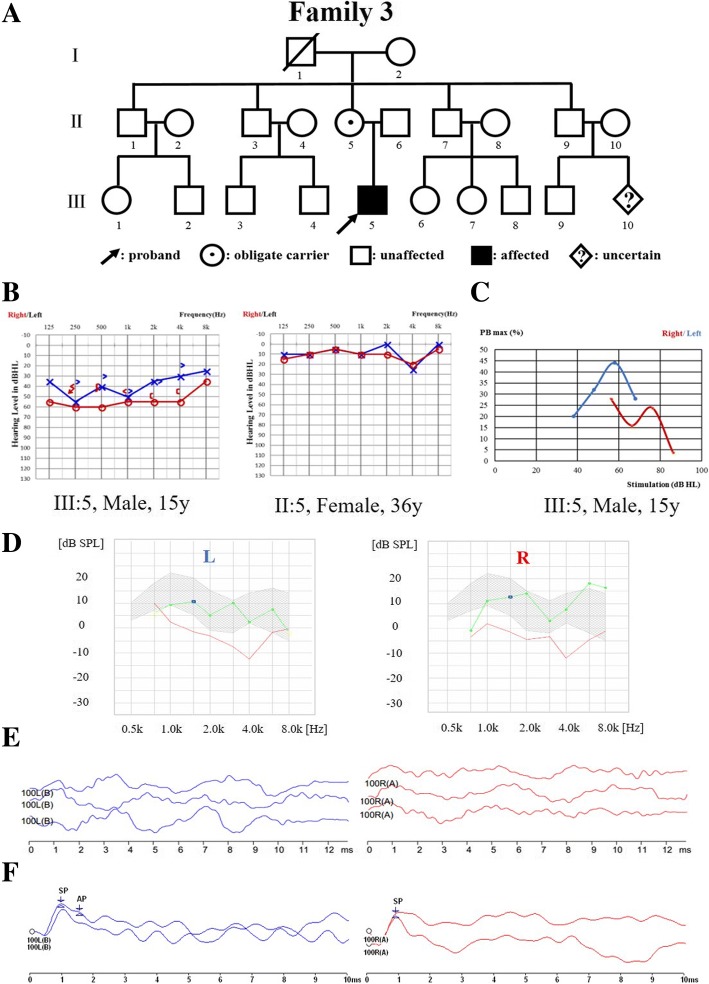
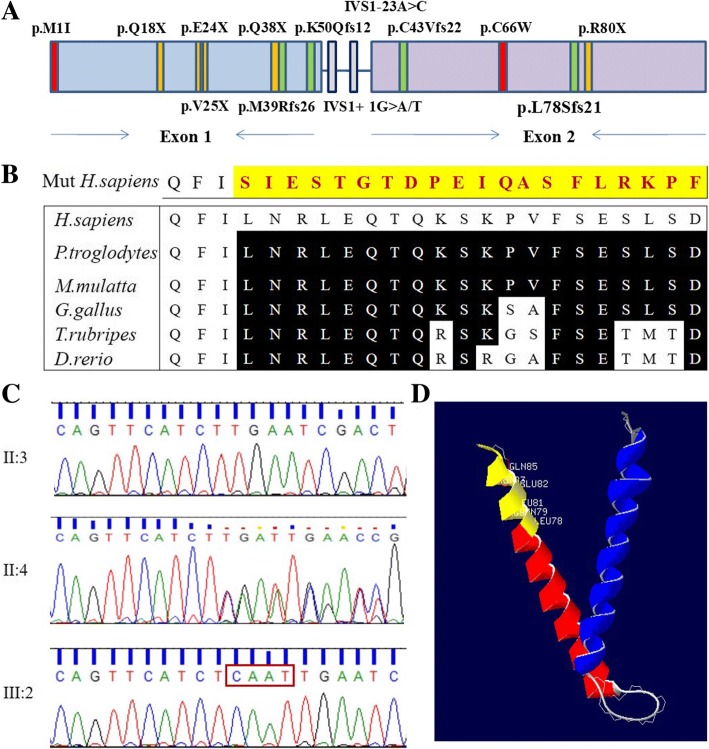
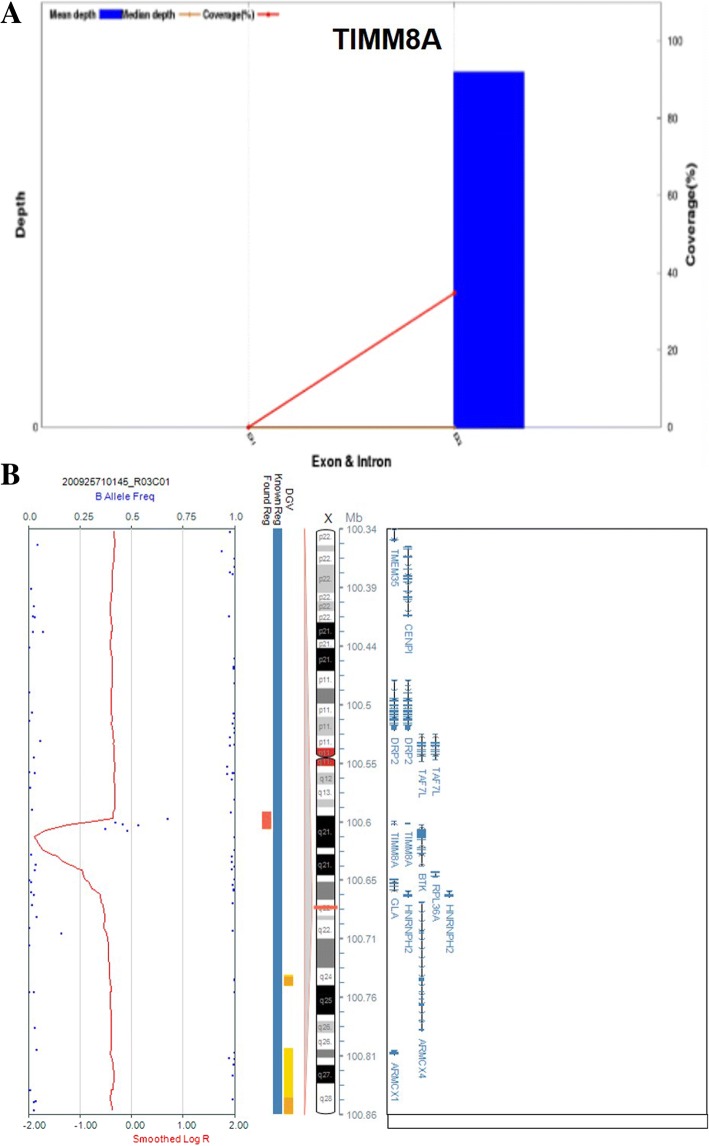
Results: A novel variation, c.232_233insCAAT, in TIMM8A was identified as the pathogenic variation in one Chinese family. This variation co-segregated with the most frequent phenotypic deafness and was absent in the 1000 Genomes Project, ExAC and 1751 ethnicity-matched controls. Clinically, otological examinations illustrated the typical postsynaptic auditory neuropathy for the proband without the symptoms of dystonia or optic atrophy. MRI demonstrated abnormal small cochlear symmetric nerves, while the vestibular function appeared to be less influenced. Furthermore, we found another two TIMM8A variations, the deletion c.133_135delGAG and a copy number variation (CNV) including the TIMM8A gene, in two independent case, when we performed NGS on an auditory neuropathy population.
Conclusion: We identified two novel variations in the TIMM8A gene (c.232_233insCAAT and c.133_135delGAG) and a CNV including the TIMM8A gene in three independent Chinese families with predicted MTS. To our knowledge, this is the first report of TIMM8A variations being identified in a Chinese population. Our results enrich the variation spectrum of TIMM8A and clinical heterogeneity of MTS. Genetic detection and diagnosis is a powerful tool for better understanding and managing syndromic hearing impairments, such as MTS, before they become full-blown.
Keywords: Auditory neuropathy; Mohr-Tranebjaerg syndrome (MTS); TIMM8A.
Conflict of interest statement
Ethics approval and consent to participate
The study was approved by the Committee of Medical Ethics of Chinese PLA General Hospital. Written informed consent from all the participants in the family was obtained.
Consent for publication
Written informed consents for publication from all the participants in the family were obtained from the next of kin on the behalf of the minors/children participants involved in this study.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Mohr J, Mageroy K. Sex-linked deafness of a possibly new type. Acta Genet Stat Med. 1960;10:54–62. - PubMed
-
- Tranebjaerg L. Deafness-dystonia-optic Neuronopathy syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, LJH B, Bird TD, Fong CT, Mefford HC, RJH S, et al., editors. GeneReviews(R). Seattle; 1993.
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
