

The Nucleotide Sensor ZBP1 and Kinase RIPK3 Induce the Enzyme IRG1 to Promote an Antiviral Metabolic State in Neurons
- PMID: 30635240
- PMCID: PMC6342485
- DOI: 10.1016/j.immuni.2018.11.017
The Nucleotide Sensor ZBP1 and Kinase RIPK3 Induce the Enzyme IRG1 to Promote an Antiviral Metabolic State in Neurons
Abstract
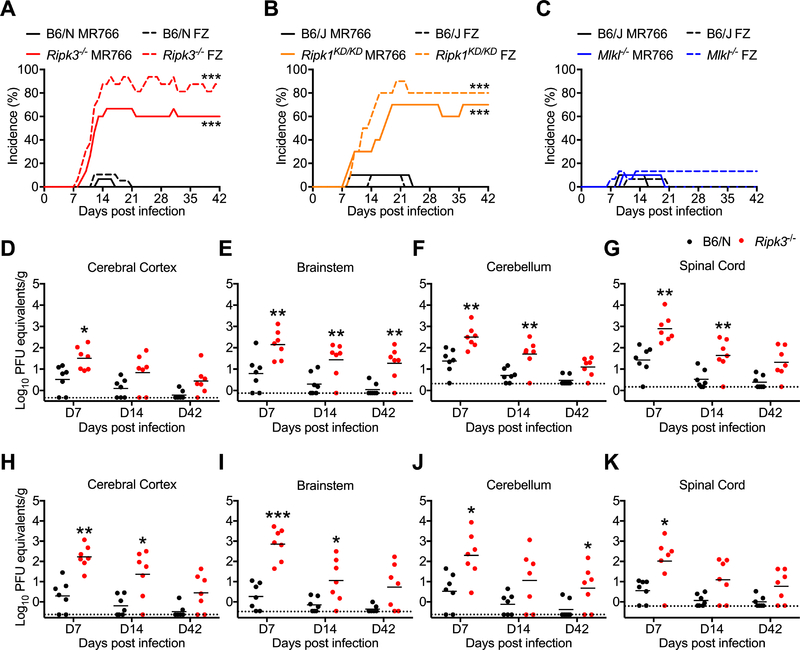
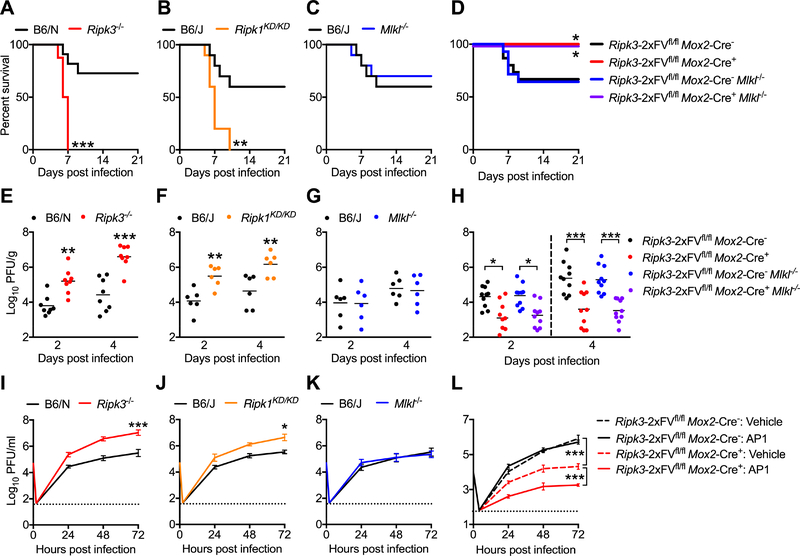
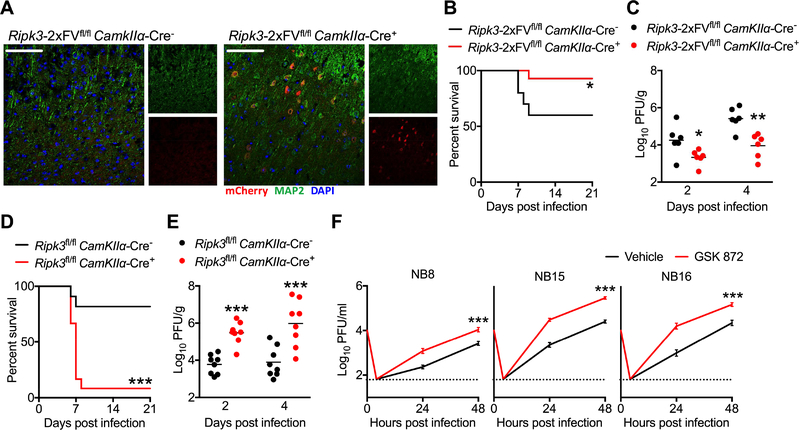
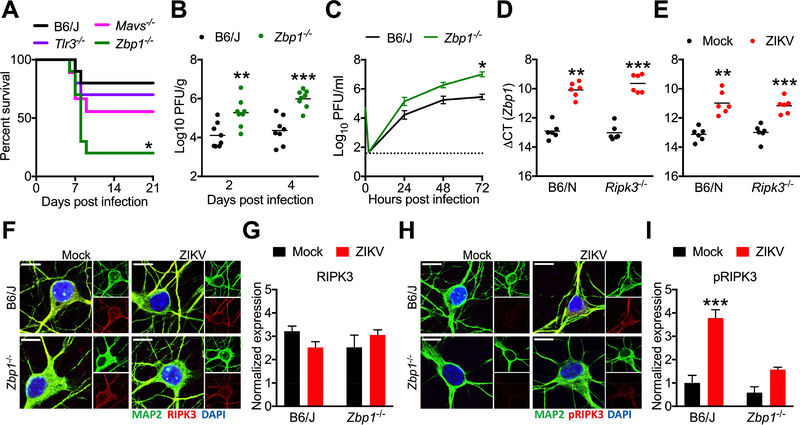
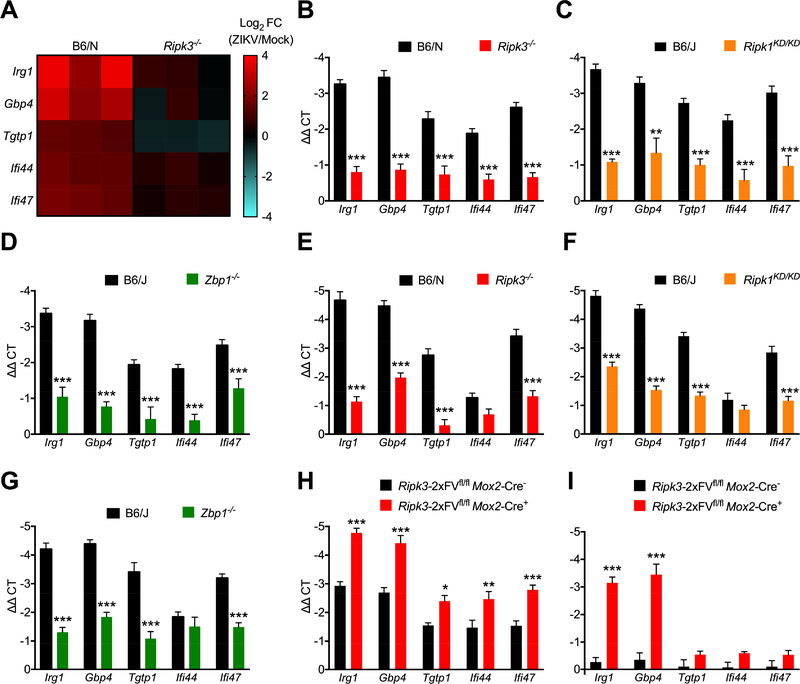
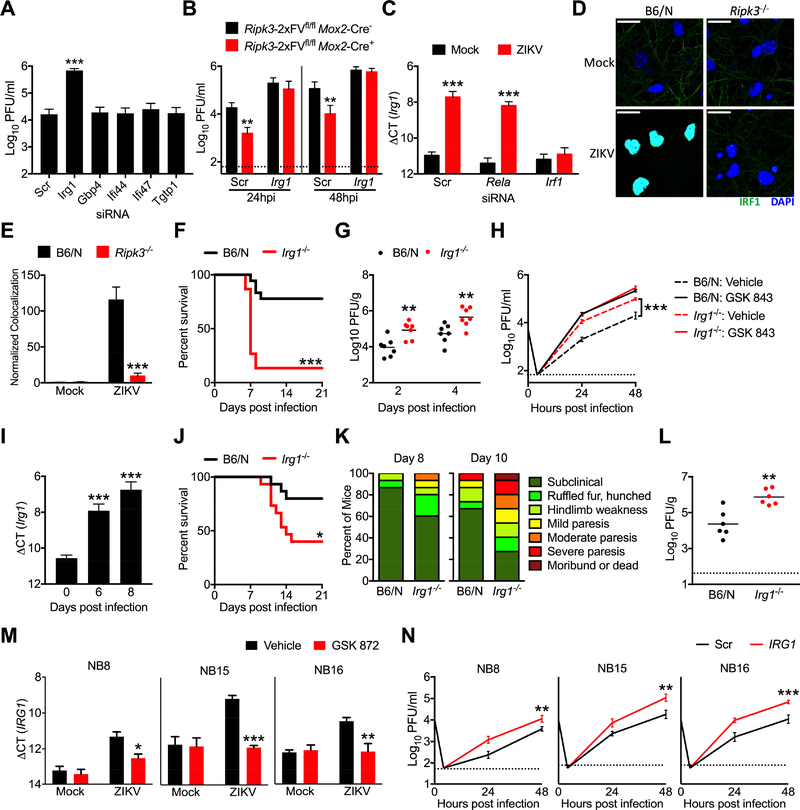
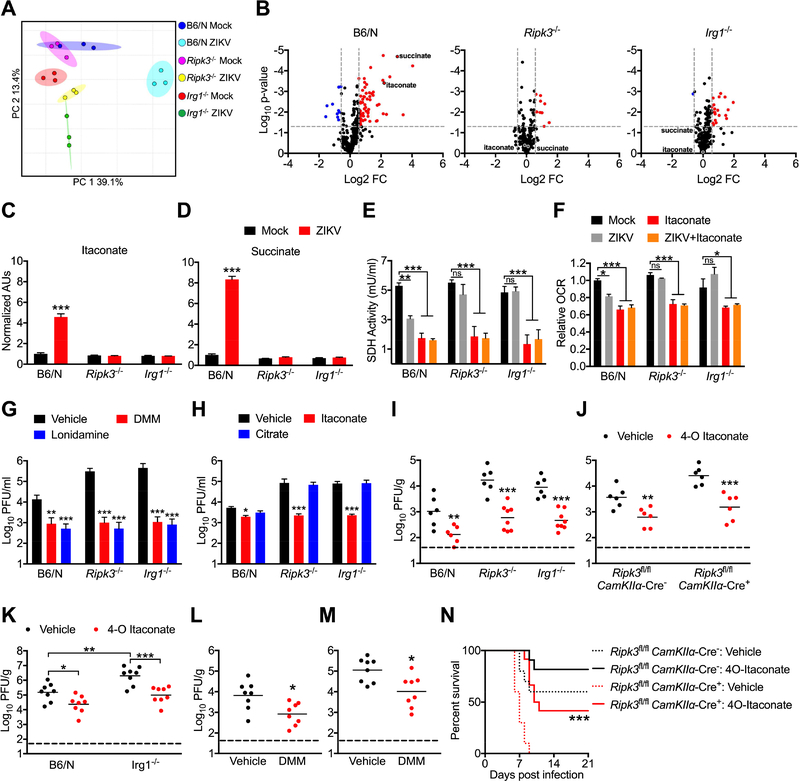
As long-lived post-mitotic cells, neurons employ unique strategies to resist pathogen infection while preserving cellular function. Here, using a murine model of Zika virus (ZIKV) infection, we identified an innate immune pathway that restricts ZIKV replication in neurons and is required for survival upon ZIKV infection of the central nervous system (CNS). We found that neuronal ZIKV infection activated the nucleotide sensor ZBP1 and the kinases RIPK1 and RIPK3, core components of virus-induced necroptotic cell death signaling. However, activation of this pathway in ZIKV-infected neurons did not induce cell death. Rather, RIPK signaling restricted viral replication by altering cellular metabolism via upregulation of the enzyme IRG1 and production of the metabolite itaconate. Itaconate inhibited the activity of succinate dehydrogenase, generating a metabolic state in neurons that suppresses replication of viral genomes. These findings demonstrate an immunometabolic mechanism of viral restriction during neuroinvasive infection.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests
Drs. Daniels and Oberst hold a provisional patent describing the use of itaconate as an antiviral molecule in the context of neuroinvasive infection.
Figures
Comment in
-
Neuronal itaconate restricts viral infection.Nat Rev Immunol. 2019 Feb;19(2):67. doi: 10.1038/s41577-019-0121-z. Nat Rev Immunol. 2019. PMID: 30644450 No abstract available.
-
RIPK3: Beyond Necroptosis.Immunity. 2019 Jan 15;50(1):1-3. doi: 10.1016/j.immuni.2018.12.031. Immunity. 2019. PMID: 30650369
References
-
- Brien JD, Lazear HM, and Diamond MS (2013). Propagation, quantification, detection, and storage of West Nile virus. Current protocols in microbiology 31, 15D 13 11–15D 13 18. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
