

Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice
- PMID: 30635357
- PMCID: PMC6363427
- DOI: 10.1084/jem.20180729
Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice
Abstract
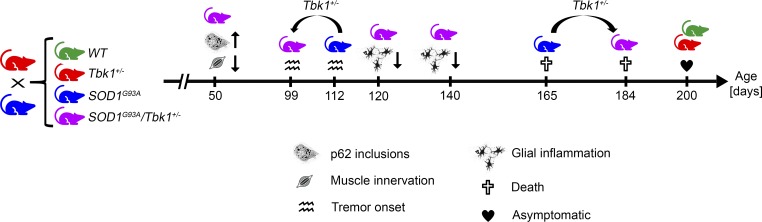
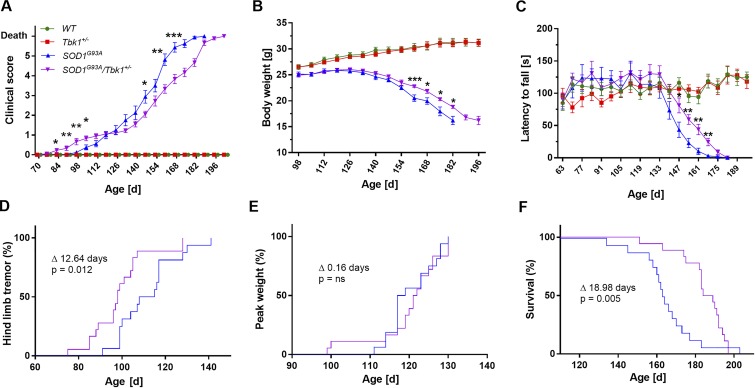
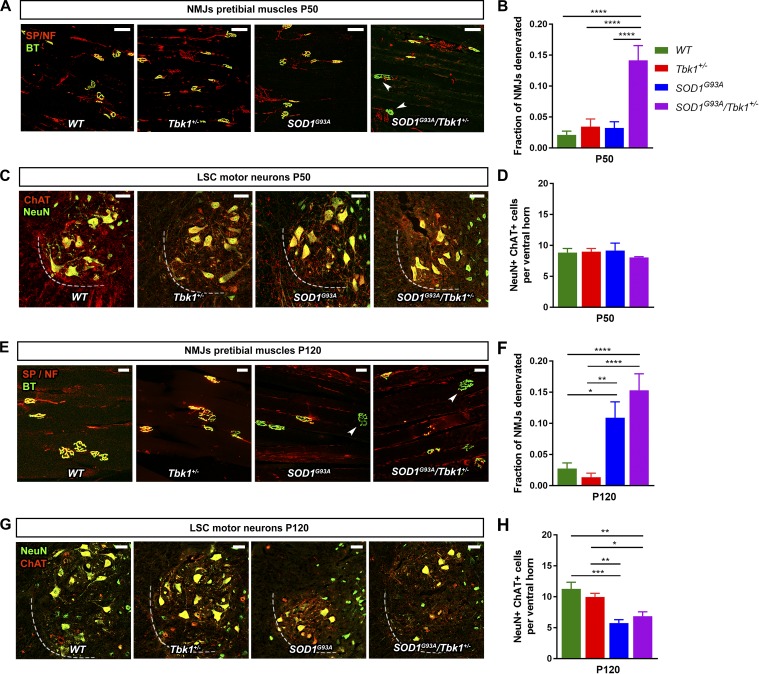
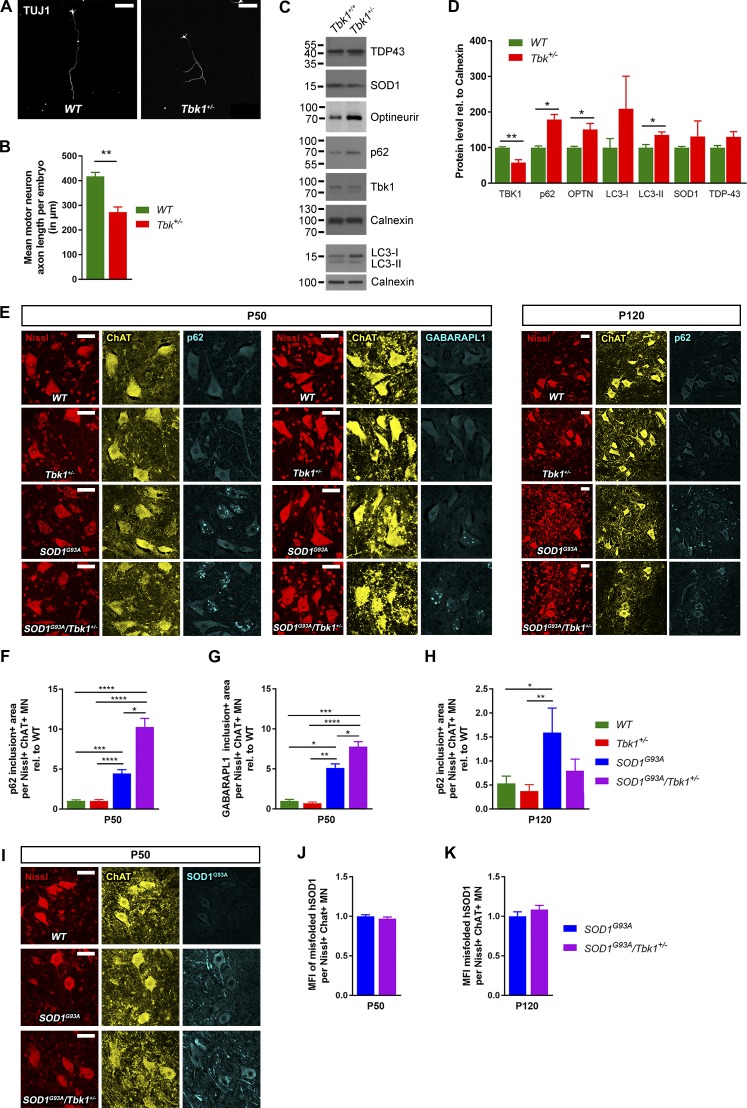
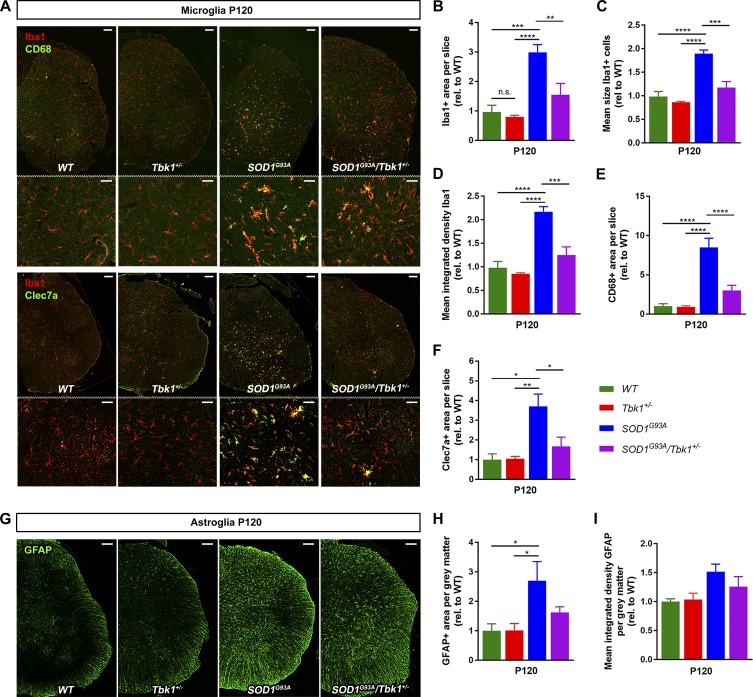
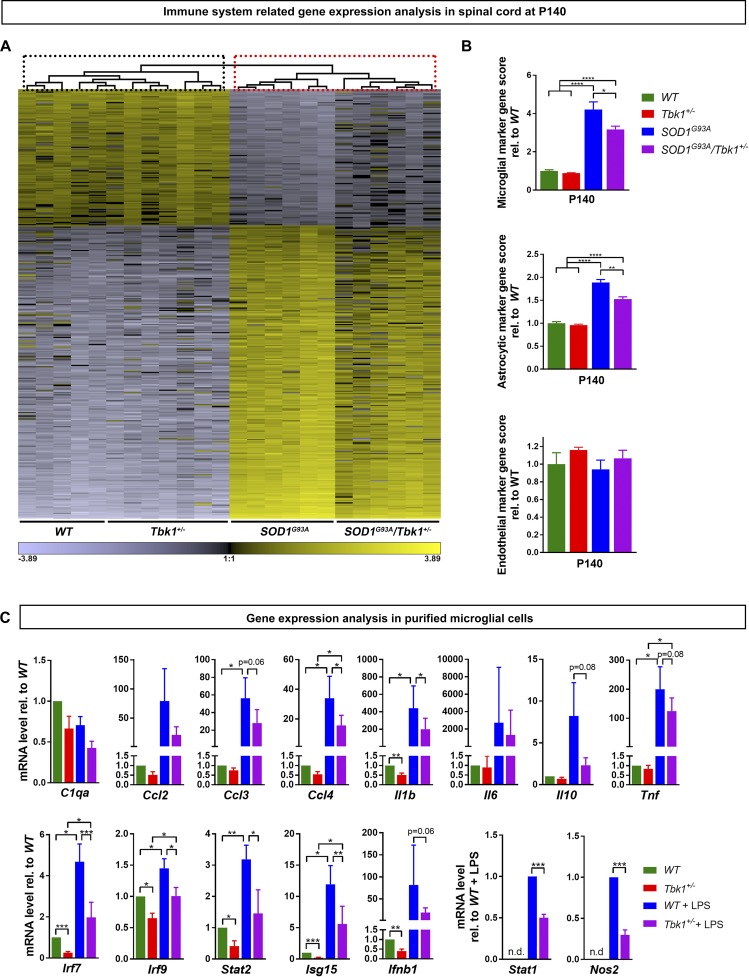
Heterozygous loss-of-function mutations of TANK-binding kinase 1 (TBK1 ) cause familial ALS, yet downstream mechanisms of TBK1 mutations remained elusive. TBK1 is a pleiotropic kinase involved in the regulation of selective autophagy and inflammation. We show that heterozygous Tbk1 deletion alone does not lead to signs of motoneuron degeneration or disturbed autophagy in mice during a 200-d observation period. Surprisingly, however, hemizygous deletion of Tbk1 inversely modulates early and late disease phases in mice additionally overexpressing ALS-linked SOD1G93A , which represents a "second hit" that induces both neuroinflammation and proteostatic dysregulation. At the early stage, heterozygous Tbk1 deletion impairs autophagy in motoneurons and prepones both the clinical onset and muscular denervation in SOD1G93A/Tbk1+/- mice. At the late disease stage, however, it significantly alleviates microglial neuroinflammation, decelerates disease progression, and extends survival. Our results indicate a profound effect of TBK1 on brain inflammatory cells under pro-inflammatory conditions and point to a complex, two-edged role of TBK1 in SOD1-linked ALS.
© 2019 Brenner et al.
Figures
References
-
- Beers D.R., Henkel J.S., Xiao Q., Zhao W., Wang J., Yen A.A., Siklos L., McKercher S.R., and Appel S.H.. 2006. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA. 103:16021–16026. 10.1073/pnas.0607423103 - DOI - PMC - PubMed
-
- Boillée S., Yamanaka K., Lobsiger C.S., Copeland N.G., Jenkins N.A., Kassiotis G., Kollias G., and Cleveland D.W.. 2006. Onset and Progression in Inherited ALS Determined by MNs and Microglia. Science. 312:1389–1392. - PubMed
-
- Bonnard M., Mirtsos C., Suzuki S., Graham K., Huang J., Ng M., Itié A., Wakeham A., Shahinian A., Henzel W.J., et al. 2000. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 19:4976–4985. 10.1093/emboj/19.18.4976 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
