

Pentatricopeptide repeat poly(A) binding protein KPAF4 stabilizes mitochondrial mRNAs in Trypanosoma brucei
- PMID: 30635574
- PMCID: PMC6329795
- DOI: 10.1038/s41467-018-08137-2
Pentatricopeptide repeat poly(A) binding protein KPAF4 stabilizes mitochondrial mRNAs in Trypanosoma brucei
Abstract
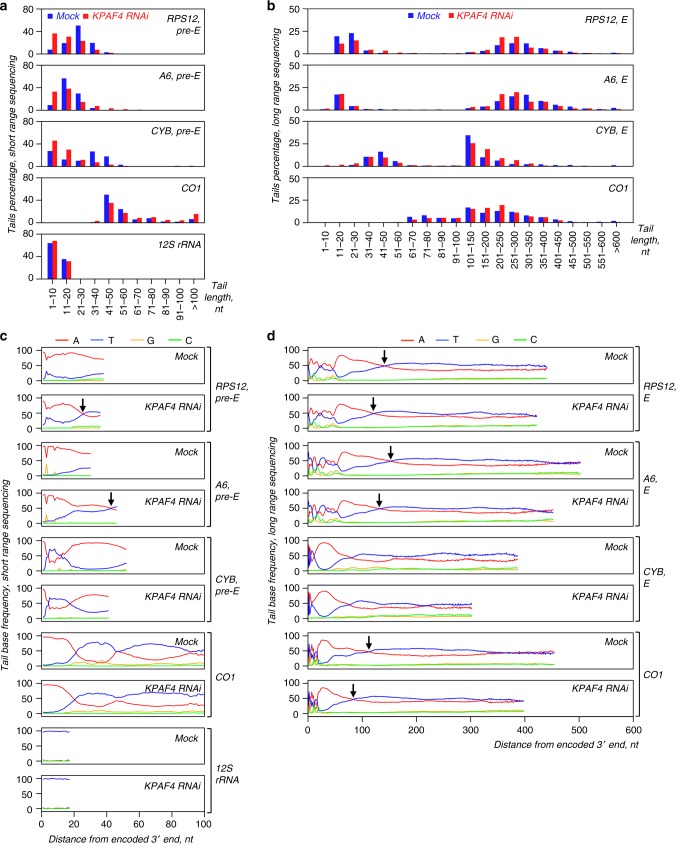
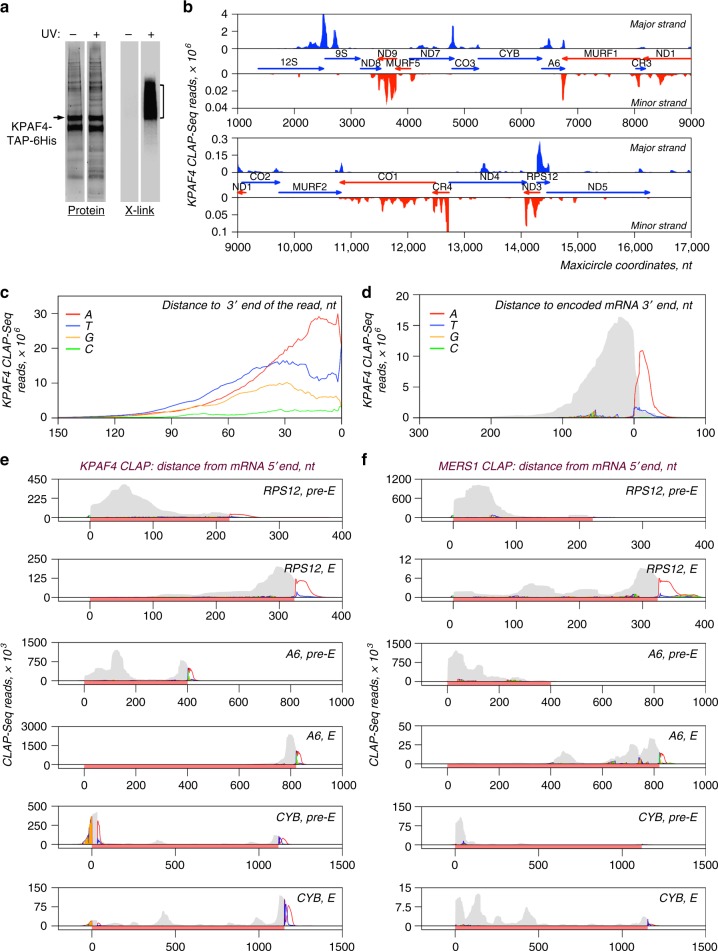
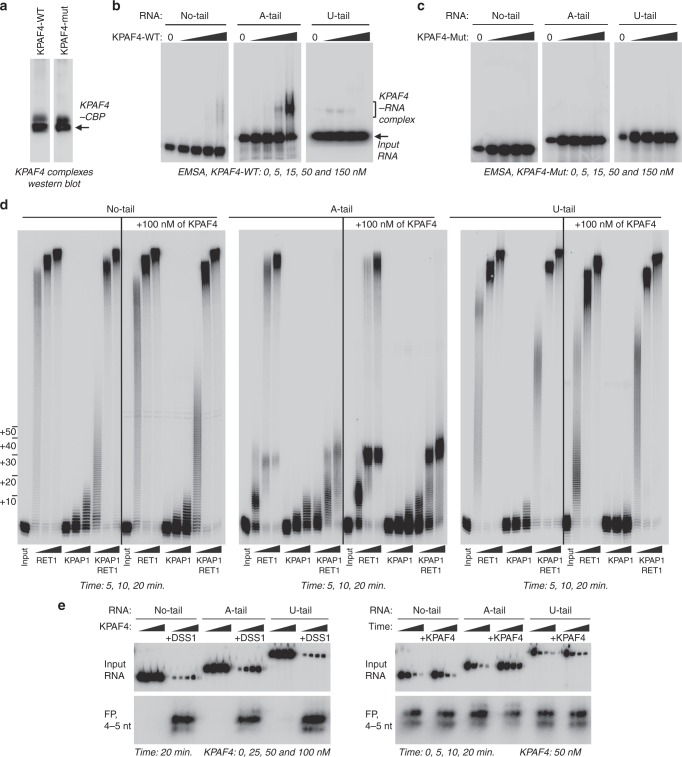
In Trypanosoma brucei, most mitochondrial mRNAs undergo editing, and 3' adenylation and uridylation. The internal sequence changes and terminal extensions are coordinated: pre-editing addition of the short (A) tail protects the edited transcript against 3'-5' degradation, while post-editing A/U-tailing renders mRNA competent for translation. Participation of a poly(A) binding protein (PABP) in coupling of editing and 3' modification processes has been inferred, but its identity and mechanism of action remained elusive. We report identification of KPAF4, a pentatricopeptide repeat-containing PABP which sequesters the A-tail and impedes mRNA degradation. Conversely, KPAF4 inhibits uridylation of A-tailed transcripts and, therefore, premature A/U-tailing of partially-edited mRNAs. This quality check point likely prevents translation of incompletely edited mRNAs. We also find that RNA editing substrate binding complex (RESC) mediates the interaction between the 5' end-bound pyrophosphohydrolase MERS1 and 3' end-associated KPAF4 to enable mRNA circularization. This event appears to be critical for edited mRNA stability.
Conflict of interest statement
The authors declare no competing interests.
Figures
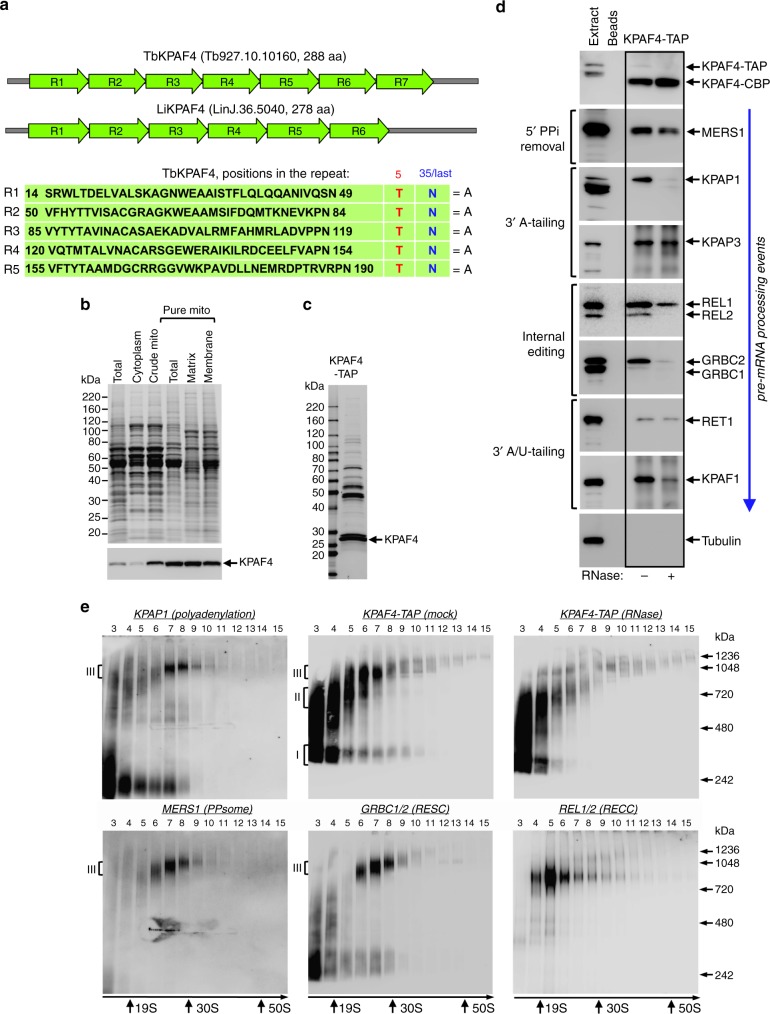
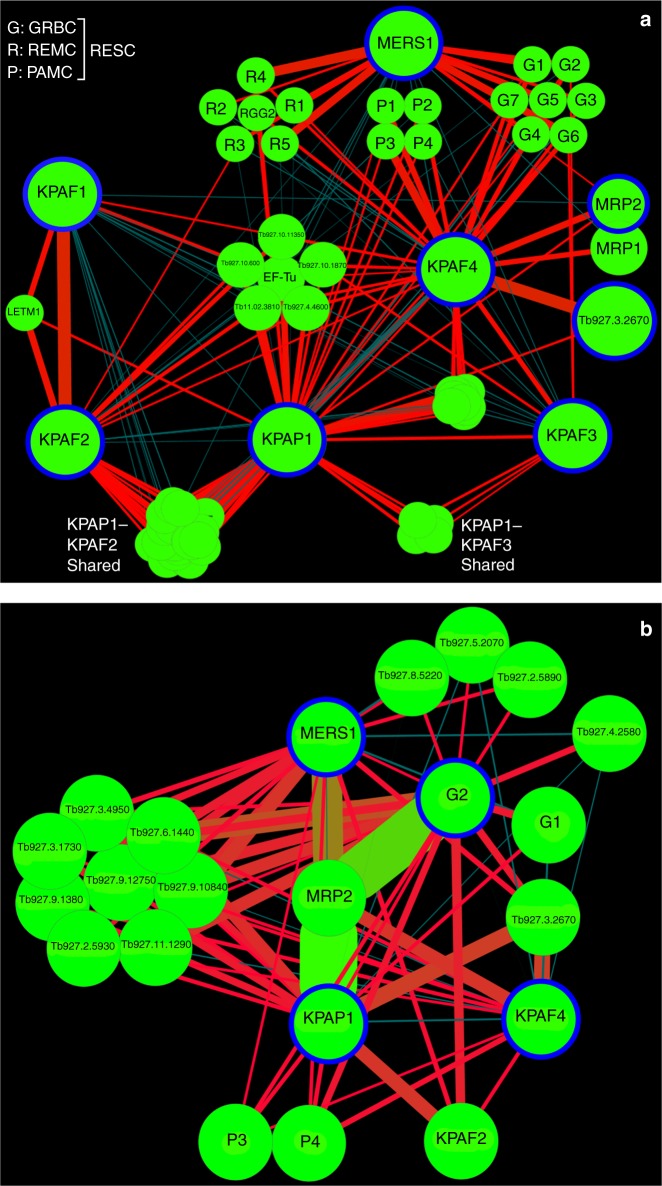
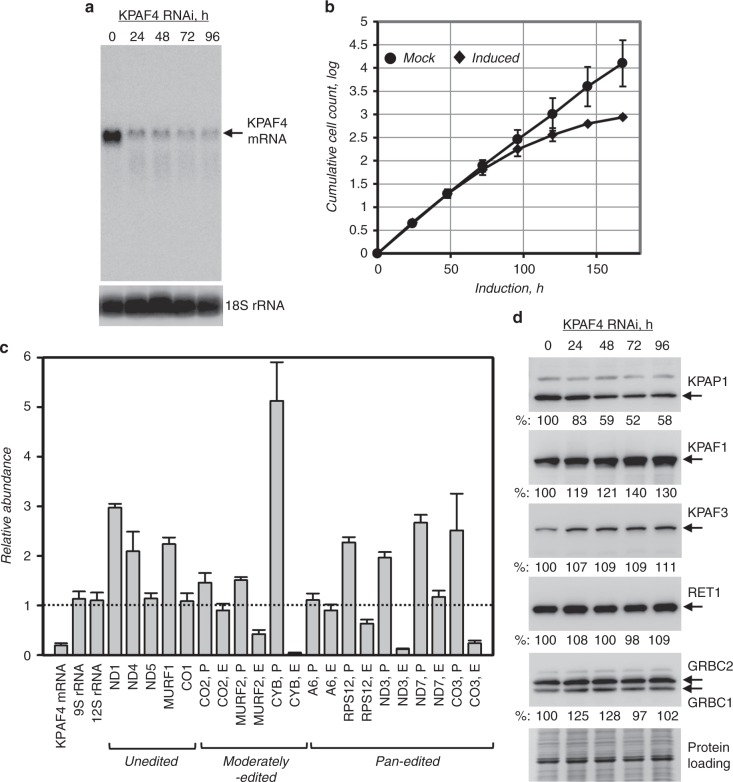
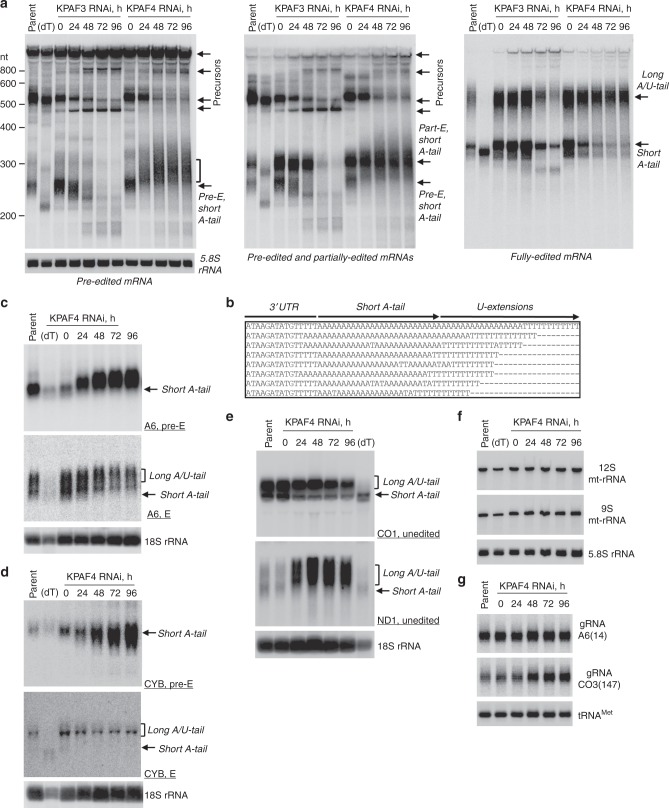
Similar articles
-
Lexis and Grammar of Mitochondrial RNA Processing in Trypanosomes.Trends Parasitol. 2020 Apr;36(4):337-355. doi: 10.1016/j.pt.2020.01.006. Epub 2020 Feb 28. Trends Parasitol. 2020. PMID: 32191849 Free PMC article. Review.
-
Poly(A) binding KPAF4/5 complex stabilizes kinetoplast mRNAs in Trypanosoma brucei.Nucleic Acids Res. 2020 Sep 4;48(15):8645-8662. doi: 10.1093/nar/gkaa575. Nucleic Acids Res. 2020. PMID: 32614436 Free PMC article.
-
Pentatricopeptide repeat proteins stimulate mRNA adenylation/uridylation to activate mitochondrial translation in trypanosomes.Mol Cell. 2011 Apr 8;42(1):106-17. doi: 10.1016/j.molcel.2011.02.021. Mol Cell. 2011. PMID: 21474072 Free PMC article.
-
Developmental regulation of edited CYb and COIII mitochondrial mRNAs is achieved by distinct mechanisms in Trypanosoma brucei.Nucleic Acids Res. 2020 Sep 4;48(15):8704-8723. doi: 10.1093/nar/gkaa641. Nucleic Acids Res. 2020. PMID: 32738044 Free PMC article.
-
Constructive edge of uridylation-induced RNA degradation.RNA Biol. 2016 Nov;13(11):1078-1083. doi: 10.1080/15476286.2016.1229736. Epub 2016 Oct 7. RNA Biol. 2016. PMID: 27715485 Free PMC article. Review.
Cited by
-
Genetic therapy in a mitochondrial disease model suggests a critical role for liver dysfunction in mortality.Elife. 2022 Nov 21;11:e65488. doi: 10.7554/eLife.65488. Elife. 2022. PMID: 36408801 Free PMC article.
-
CTS tag-based methods for investigating mitochondrial RNA modification factors in Trypanosoma brucei.Methods Enzymol. 2021;658:83-109. doi: 10.1016/bs.mie.2021.06.004. Epub 2021 Jul 14. Methods Enzymol. 2021. PMID: 34517961 Free PMC article.
-
Circular mitochondrial-encoded mRNAs are a distinct subpopulation of mitochondrial mRNA in Trypanosoma brucei.bioRxiv [Preprint]. 2023 Feb 10:2023.02.10.528059. doi: 10.1101/2023.02.10.528059. bioRxiv. 2023. Update in: Sci Rep. 2023 May 15;13(1):7825. doi: 10.1038/s41598-023-34255-z. PMID: 36798374 Free PMC article. Updated. Preprint.
-
Lexis and Grammar of Mitochondrial RNA Processing in Trypanosomes.Trends Parasitol. 2020 Apr;36(4):337-355. doi: 10.1016/j.pt.2020.01.006. Epub 2020 Feb 28. Trends Parasitol. 2020. PMID: 32191849 Free PMC article. Review.
-
RNA editing catalytic complexes edit multiple mRNA sites non-processively in Trypanosoma brucei.Mol Biochem Parasitol. 2023 Dec;256:111596. doi: 10.1016/j.molbiopara.2023.111596. Epub 2023 Sep 22. Mol Biochem Parasitol. 2023. PMID: 37742784 Free PMC article.
