

Inhibition of cardiomyocyte Sprouty1 protects from cardiac ischemia-reperfusion injury
- PMID: 30635790
- PMCID: PMC6329741
- DOI: 10.1007/s00395-018-0713-y
Inhibition of cardiomyocyte Sprouty1 protects from cardiac ischemia-reperfusion injury
Abstract
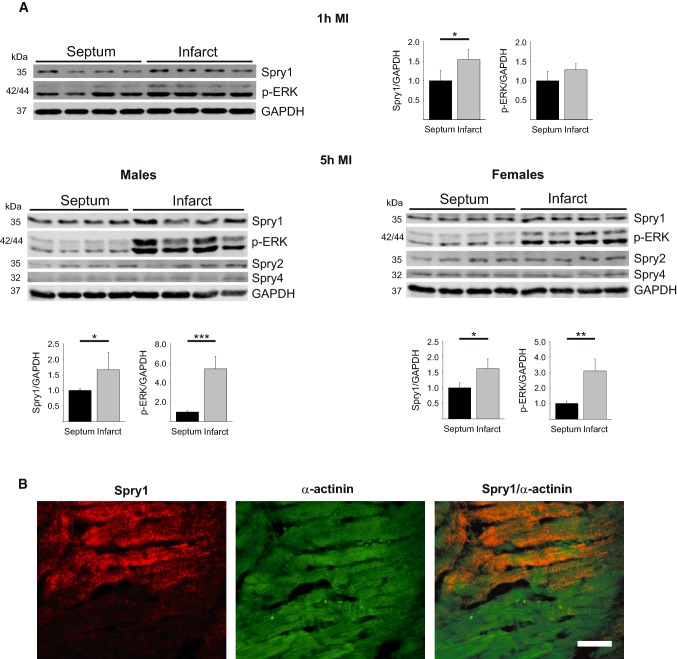
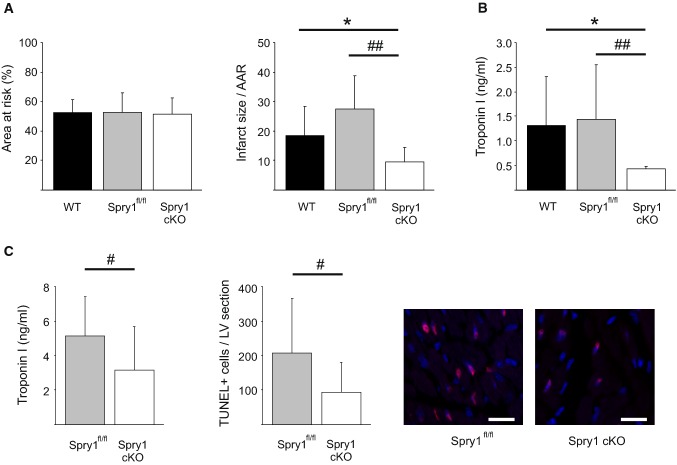
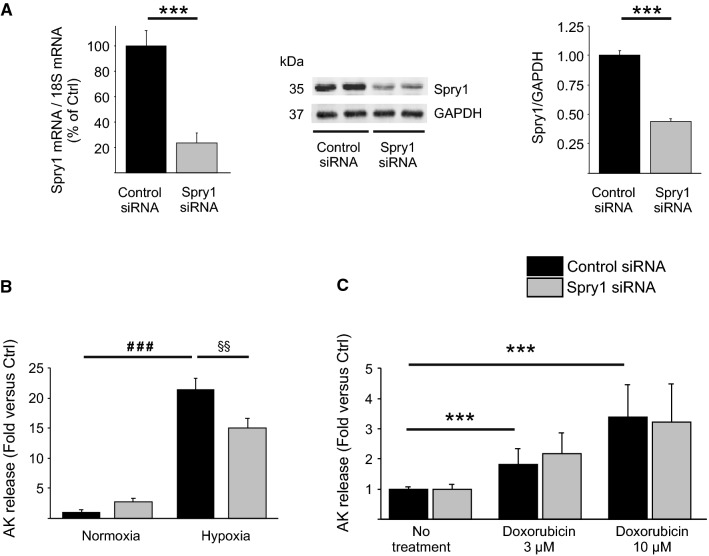
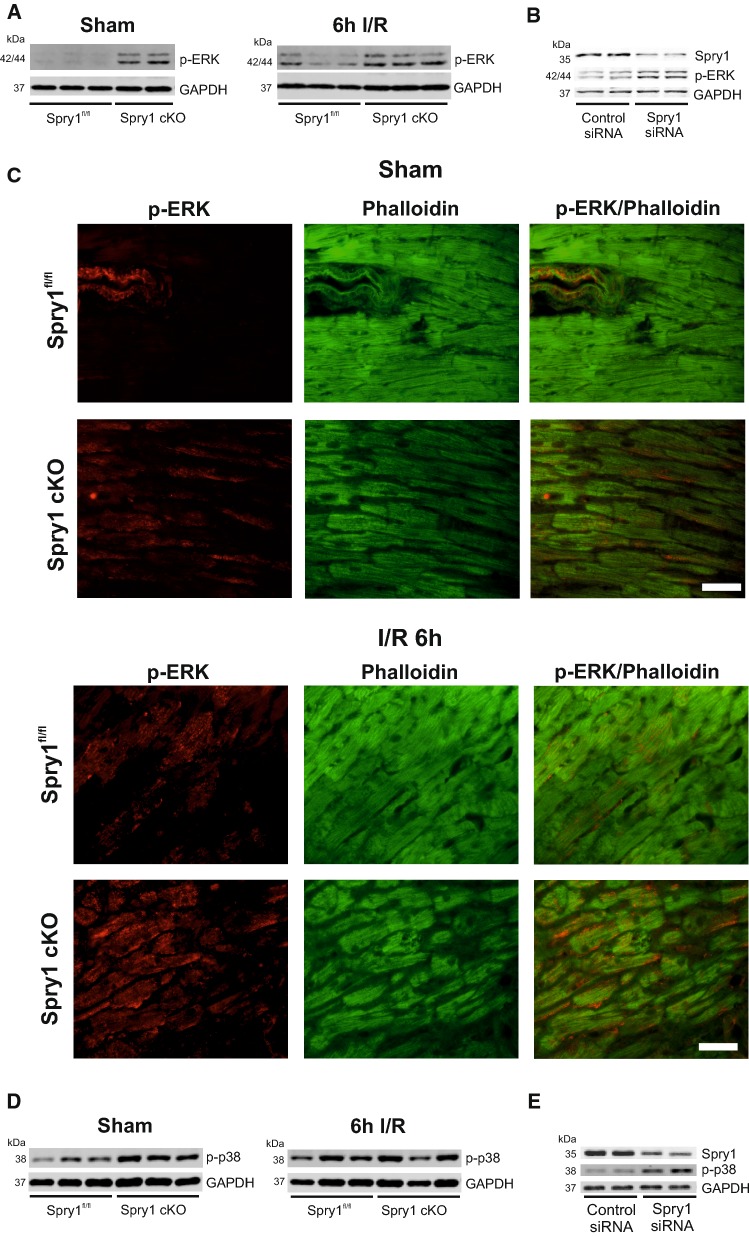
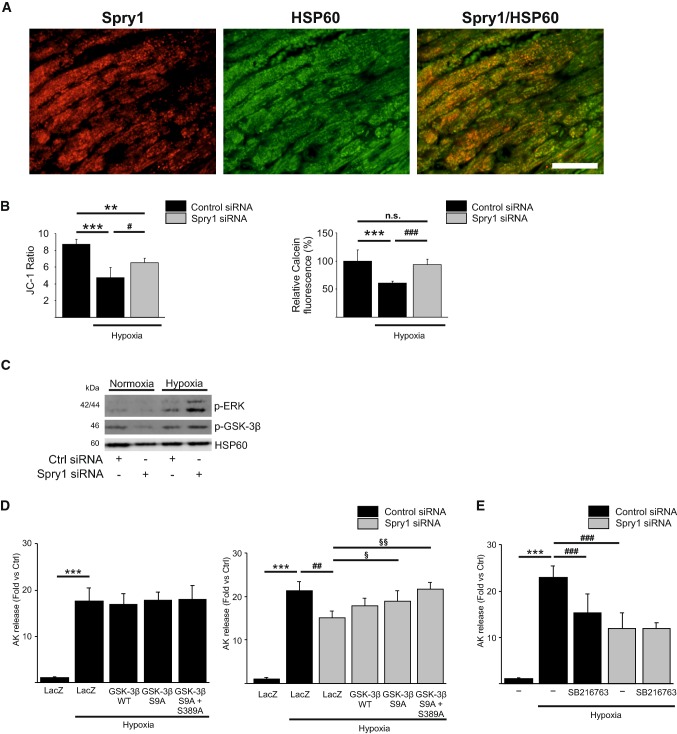
Sprouty1 (Spry1) is a negative modulator of receptor tyrosine kinase signaling, but its role in cardiomyocyte survival has not been elucidated. The aim of this study was to investigate the potential role of cardiomyocyte Spry1 in cardiac ischemia-reperfusion (I/R) injury. Infarct areas of mouse hearts showed an increase in Spry1 protein expression, which localized to cardiomyocytes. To investigate if cardiomyocyte Spry1 regulates I/R injury, 8-week-old inducible cardiomyocyte Spry1 knockout (Spry1 cKO) mice and control mice were subjected to cardiac I/R injury. Spry1 cKO mice showed reduction in release of cardiac troponin I and reduced infarct size after I/R injury compared to control mice. Similar to Spry1 knockdown in cardiomyocytes in vivo, RNAi-mediated Spry1 silencing in isolated cardiomyocytes improved cardiomyocyte survival following simulated ischemia injury. Mechanistically, Spry1 knockdown induced cardiomyocyte extracellular signal-regulated kinase (ERK) phosphorylation in healthy hearts and isolated cardiomyocytes, and enhanced ERK phosphorylation after I/R injury. Spry1-deficient cardiomyocytes showed better preserved mitochondrial membrane potential following ischemic injury and an increase in levels of phosphorylated ERK and phosphorylated glycogen synthase kinase-3β (GSK-3β) in mitochondria of hypoxic cardiomyocytes. Overexpression of constitutively active GSK-3β abrogated the protective effect of Spry1 knockdown. Moreover, pharmacological inhibition of GSK-3β protected wild-type cardiomyocytes from cell death, but did not further protect Spry1-silenced cardiomyocytes from hypoxia-induced injury. Cardiomyocyte Spry1 knockdown promotes ERK phosphorylation and offers protection from I/R injury. Our findings indicate that Spry1 is an important regulator of cardiomyocyte viability during ischemia-reperfusion injury.
Keywords: Extracellular signal-regulated kinase; Glycogen synthase kinase-3β; Ischemia–reperfusion injury; Myocardial infarction; Sprouty1.
Conflict of interest statement
The authors have no conflict of interest.
Figures
References
-
- Basson MA, Akbulut S, Watson-Johnson J, Simon R, Carroll TJ, Shakya R, Gross I, Martin GR, Lufkin T, McMahon AP, Wilson PD, Costantini FD, Mason IJ, Licht JD. Sprouty1 is a critical regulator of GDNF/RET-mediated kidney induction. Dev Cell. 2005;8:229–239. doi: 10.1016/j.devcel.2004.12.004. - DOI - PubMed
-
- Botker HE, Hausenloy D, Andreadou I, Antonucci S, Boengler K, Davidson SM, Deshwal S, Devaux Y, Di Lisa F, Di Sante M, Efentakis P, Femmino S, Garcia-Dorado D, Giricz Z, Ibanez B, Iliodromitis E, Kaludercic N, Kleinbongard P, Neuhauser M, Ovize M, Pagliaro P, Rahbek-Schmidt M, Ruiz-Meana M, Schluter KD, Schulz R, Skyschally A, Wilder C, Yellon DM, Ferdinandy P, Heusch G. Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res Cardiol. 2018;113:39. doi: 10.1007/s00395-018-0696-8. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
