

LRH1-driven transcription factor circuitry for hepatocyte identity: Super-enhancer cistromic analysis
- PMID: 30638865
- PMCID: PMC6413675
- DOI: 10.1016/j.ebiom.2018.12.056
LRH1-driven transcription factor circuitry for hepatocyte identity: Super-enhancer cistromic analysis
Abstract
Background: The injured liver loses normal function, with concomitant decrease of key identity genes. Super-enhancers contribute to mammalian cell identity. Here, we identified core transcription factors (TFs) that are active in hepatocytes, using genome-wide analysis and hierarchical ordering of super-enhancer distribution.
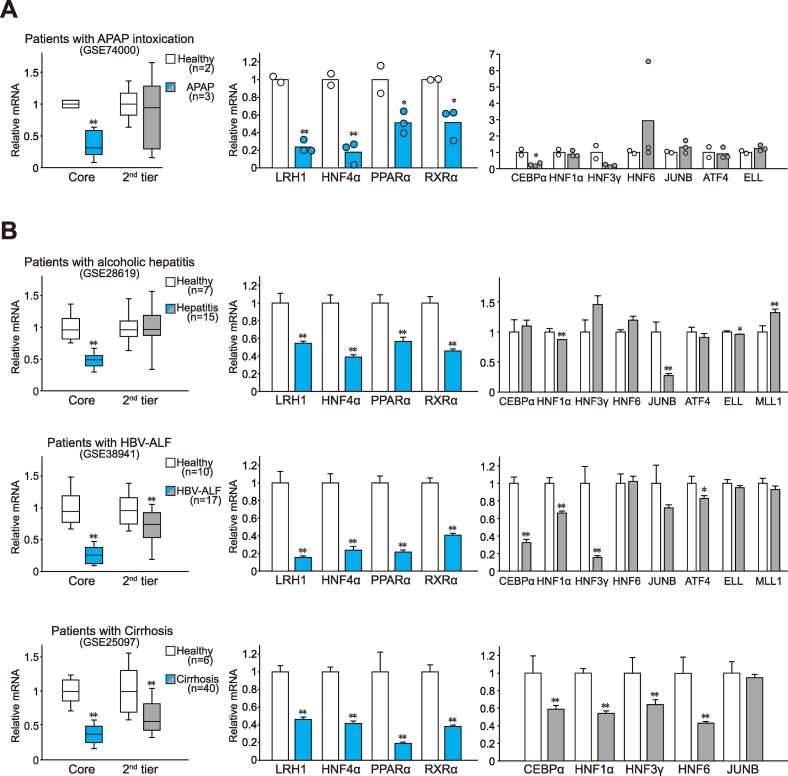
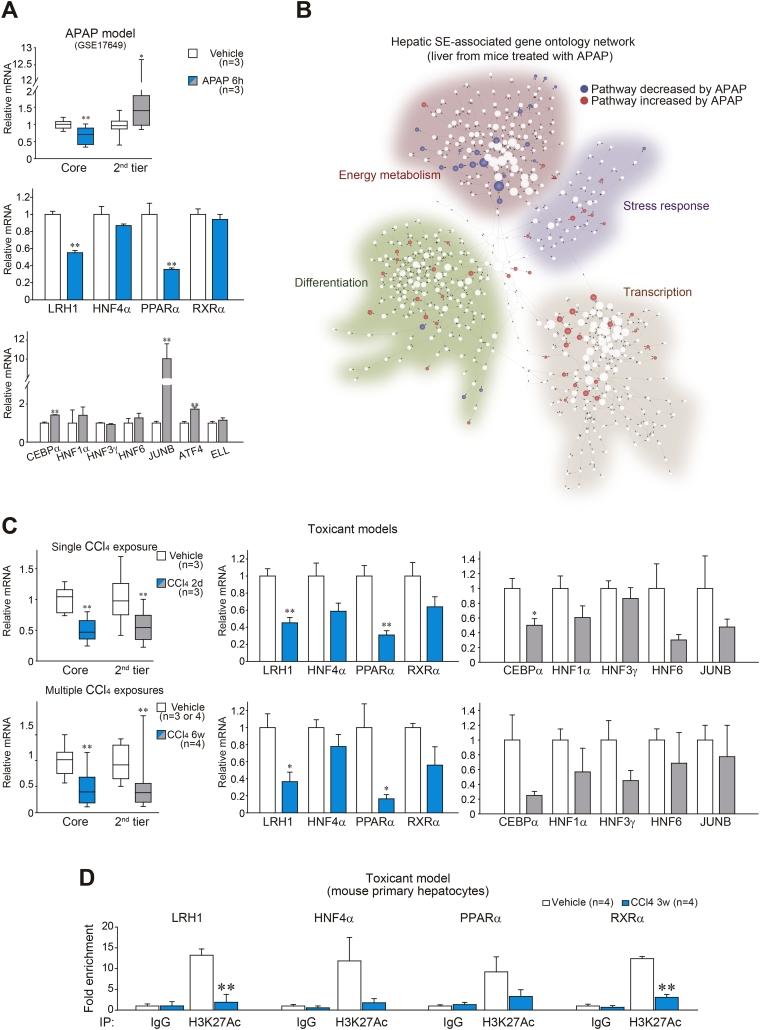
Methods: Expression of core TFs was assessed in a cohort of patients with hepatitis or cirrhosis and animal models. Quantitative PCR, chromatin immunoprecipitation assays, and hydrodynamic gene delivery methods were used to assess gene regulation and hepatocyte viability. RNA-sequencing data were generated to investigate the role of LRH1 in hepatocyte protection from injury.
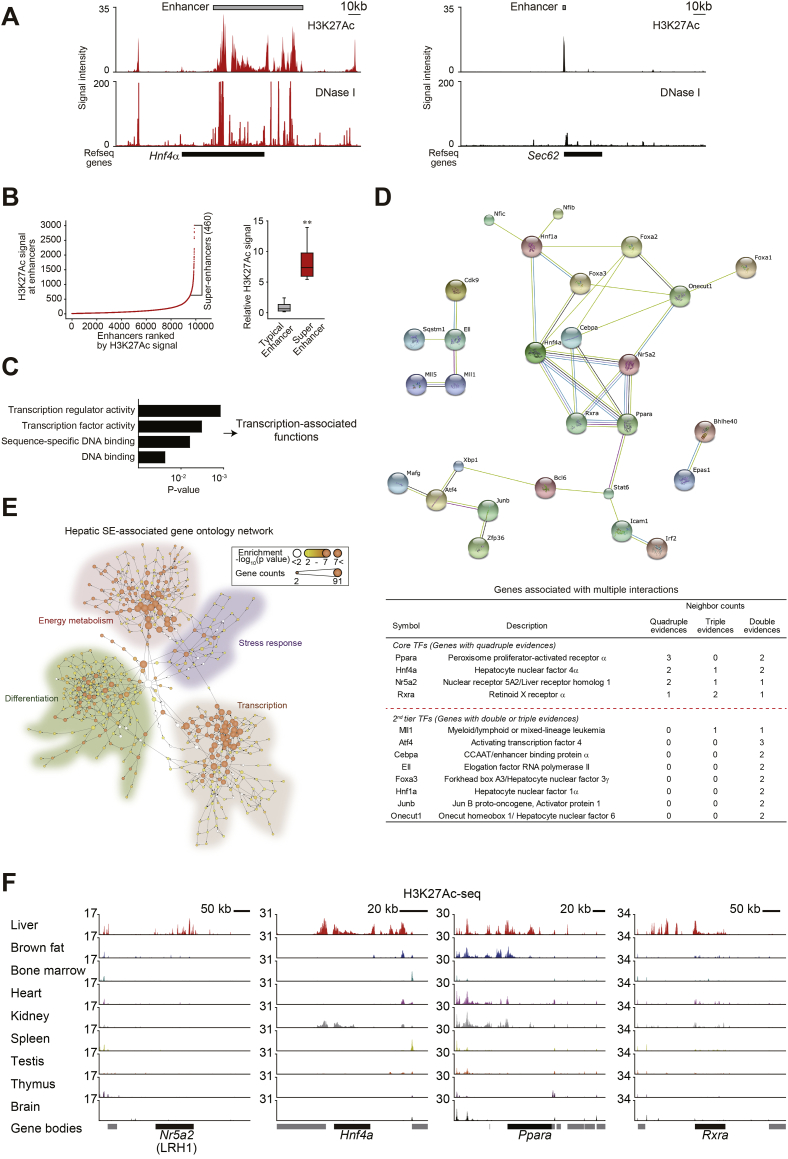
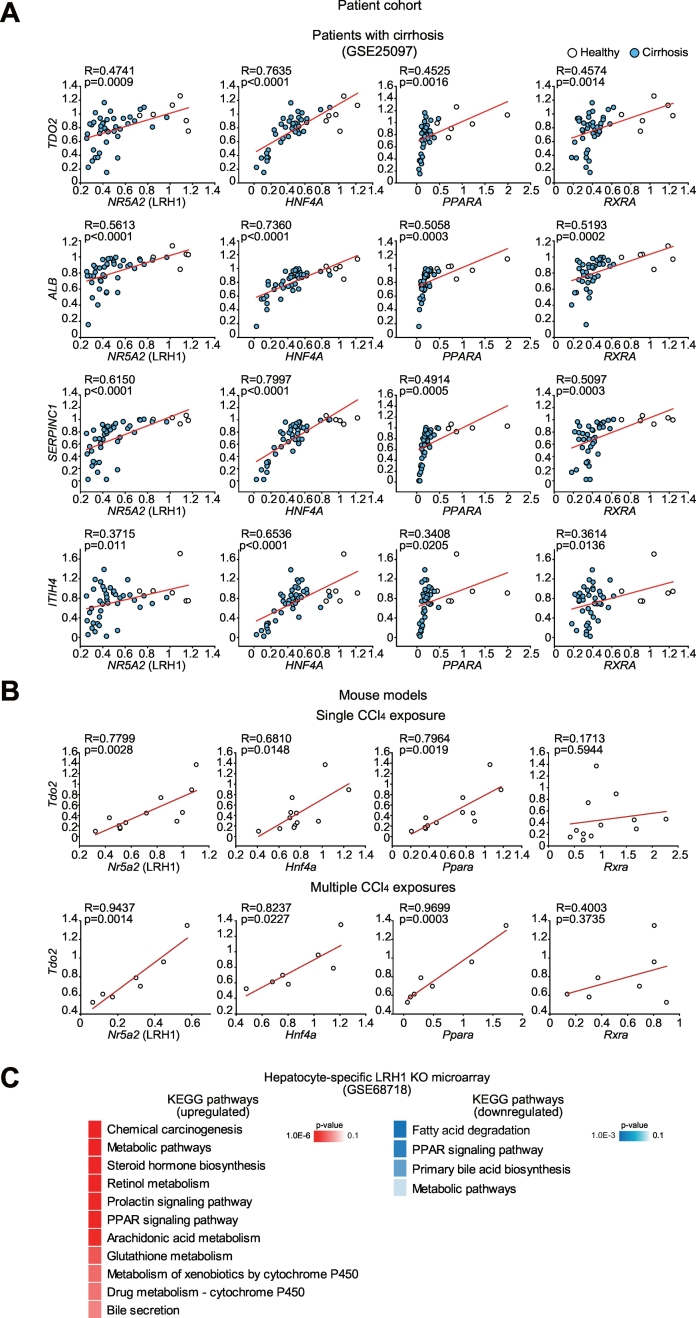
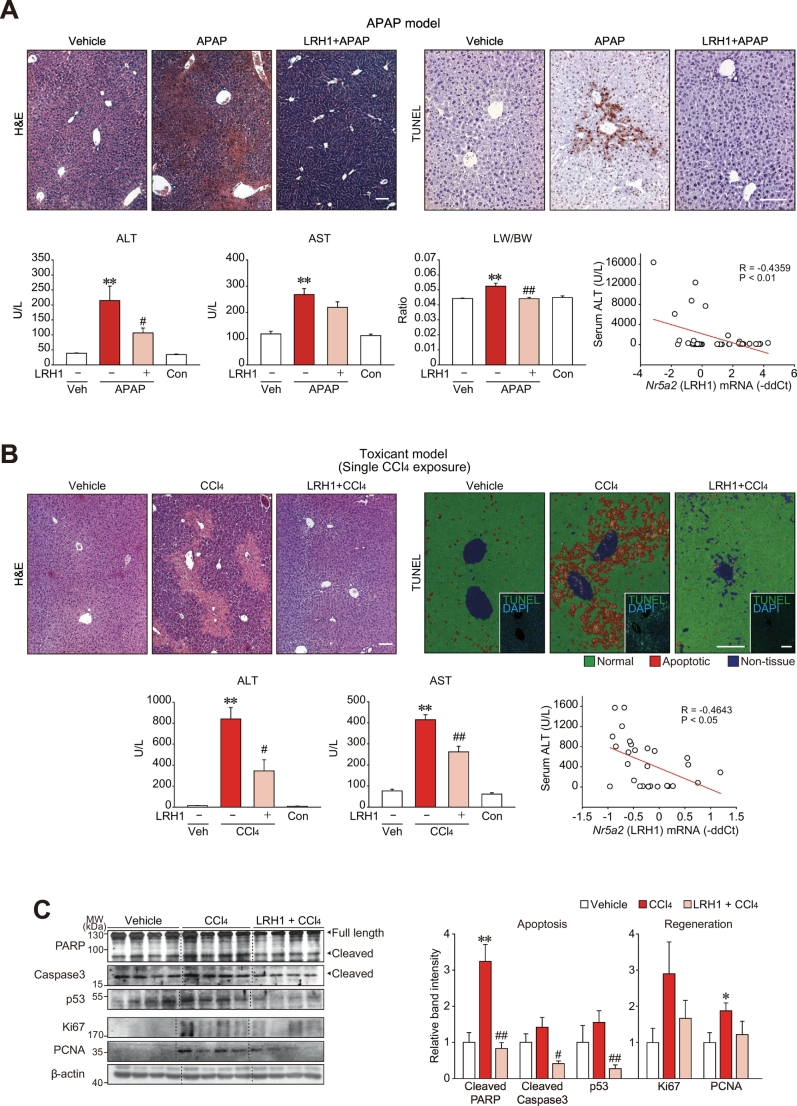
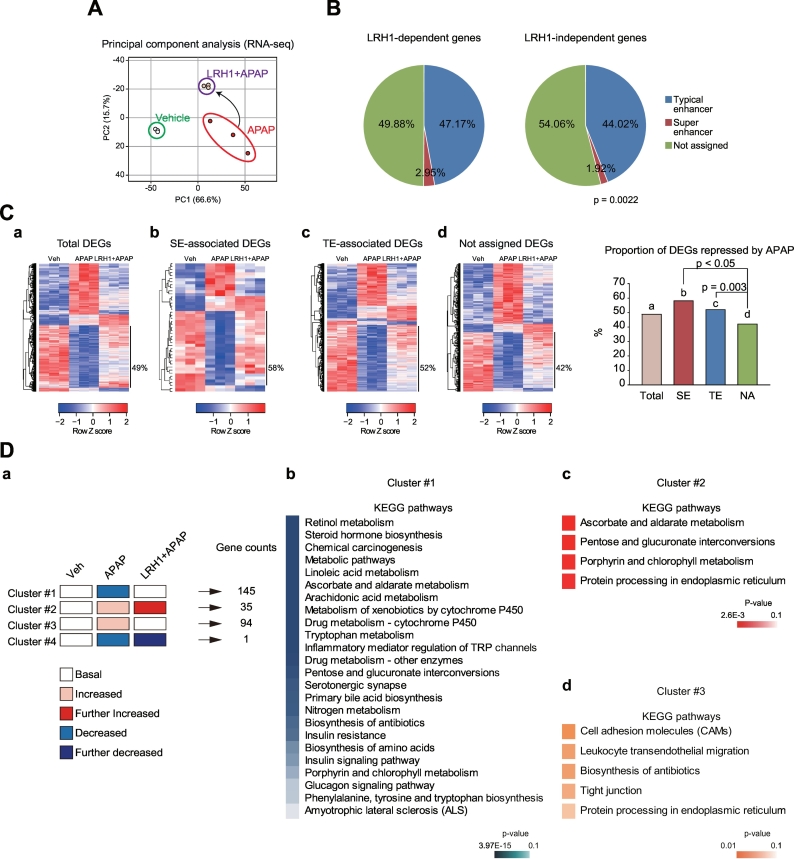
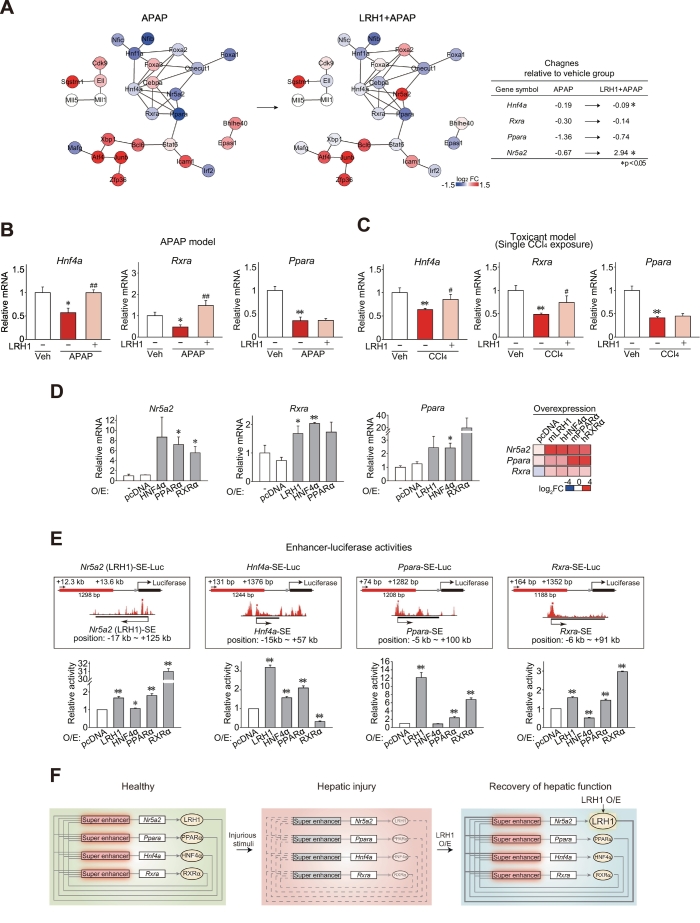
Results: Network analysis of super-enhancer-associated gene interactions and expression arrays for cohorts of patients with hepatitis and cirrhosis enabled us to identify a super-enhancer-associated network, and LRH1, HNF4α, PPARα, and RXRα as core TFs. In mouse models, expression of core TFs was robustly inhibited by single and multiple challenge(s) with liver toxicant. RNA-seq analysis revealed changes in expression in the super-enhancer-associated genes sensitively biased toward repression by intoxication. LRH1 gene delivery prevented the loss of hepatic super-enhancer-associated signaling circuitry in toxicant-challenged mice, and protected the liver from injury, indicating the role of LRH1 in hepatocyte identity and viability. In hepatocytes, overexpression of each core TF promoted induction of other TFs.
Conclusion: Overall, this study identified LRH1-driven pathway as a circuitry responsible for hepatocyte identity by using cistromic analysis, improving our understanding of liver pathophysiology and identifying novel therapeutic targets.
Keywords: Acetaminophen; Acute liver injury; LRH1; Liver disease; Super-enhancer.
Copyright © 2018. Published by Elsevier B.V.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
