

Efficient Variant Set Mixed Model Association Tests for Continuous and Binary Traits in Large-Scale Whole-Genome Sequencing Studies
- PMID: 30639324
- PMCID: PMC6372261
- DOI: 10.1016/j.ajhg.2018.12.012
Efficient Variant Set Mixed Model Association Tests for Continuous and Binary Traits in Large-Scale Whole-Genome Sequencing Studies
Abstract
With advances in whole-genome sequencing (WGS) technology, more advanced statistical methods for testing genetic association with rare variants are being developed. Methods in which variants are grouped for analysis are also known as variant-set, gene-based, and aggregate unit tests. The burden test and sequence kernel association test (SKAT) are two widely used variant-set tests, which were originally developed for samples of unrelated individuals and later have been extended to family data with known pedigree structures. However, computationally efficient and powerful variant-set tests are needed to make analyses tractable in large-scale WGS studies with complex study samples. In this paper, we propose the variant-set mixed model association tests (SMMAT) for continuous and binary traits using the generalized linear mixed model framework. These tests can be applied to large-scale WGS studies involving samples with population structure and relatedness, such as in the National Heart, Lung, and Blood Institute's Trans-Omics for Precision Medicine (TOPMed) program. SMMATs share the same null model for different variant sets, and a virtue of this null model, which includes covariates only, is that it needs to be fit only once for all tests in each genome-wide analysis. Simulation studies show that all the proposed SMMATs correctly control type I error rates for both continuous and binary traits in the presence of population structure and relatedness. We also illustrate our tests in a real data example of analysis of plasma fibrinogen levels in the TOPMed program (n = 23,763), using the Analysis Commons, a cloud-based computing platform.
Keywords: TOPMed; generalized linear mixed model; population structure; rare variants; relatedness; variant set association test; whole-genome sequencing.
Copyright © 2018 American Society of Human Genetics. All rights reserved.
Figures
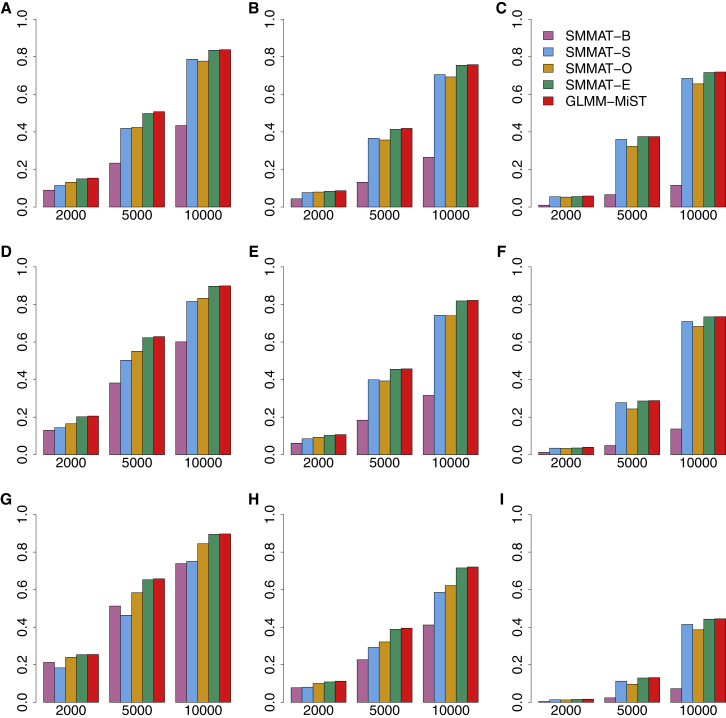
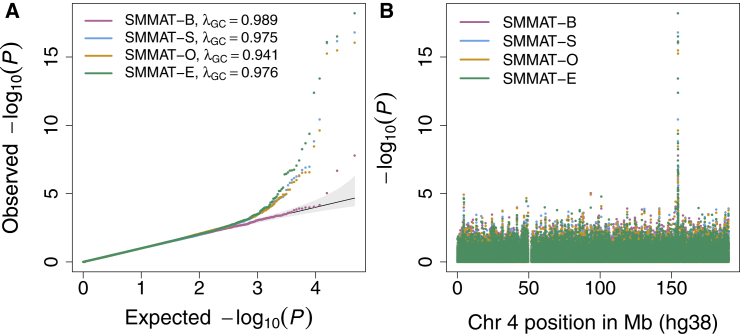
References
-
- Lippert C., Listgarten J., Liu Y., Kadie C.M., Davidson R.I., Heckerman D. FaST linear mixed models for genome-wide association studies. Nat. Methods. 2011;8:833–835. - PubMed
-
- Pirinen M., Donnelly P., Spencer C.C.A. Efficient computation with a linear mixed model on large-scale data sets with applications to genetic studies. Ann. Appl. Stat. 2013;7:369–390.
Publication types
MeSH terms
Substances
Grants and funding
- R35 CA197449/CA/NCI NIH HHS/United States
- R01 HL131136/HL/NHLBI NIH HHS/United States
- U19 CA203654/CA/NCI NIH HHS/United States
- R01 HL139553/HL/NHLBI NIH HHS/United States
- R01 HL137922/HL/NHLBI NIH HHS/United States
- R01 HL119443/HL/NHLBI NIH HHS/United States
- R35 HL135818/HL/NHLBI NIH HHS/United States
- U01 HL137162/HL/NHLBI NIH HHS/United States
- P01 CA134294/CA/NCI NIH HHS/United States
- U01 HG009088/HG/NHGRI NIH HHS/United States
- P20 GM121334/GM/NIGMS NIH HHS/United States
- R00 HL130593/HL/NHLBI NIH HHS/United States
- U54 GM115428/GM/NIGMS NIH HHS/United States
- K01 HL135405/HL/NHLBI NIH HHS/United States
- U01 HL120393/HL/NHLBI NIH HHS/United States
- R01 HL113338/HL/NHLBI NIH HHS/United States
