

Estimating recent migration and population-size surfaces
- PMID: 30640906
- PMCID: PMC6347299
- DOI: 10.1371/journal.pgen.1007908
Estimating recent migration and population-size surfaces
Abstract
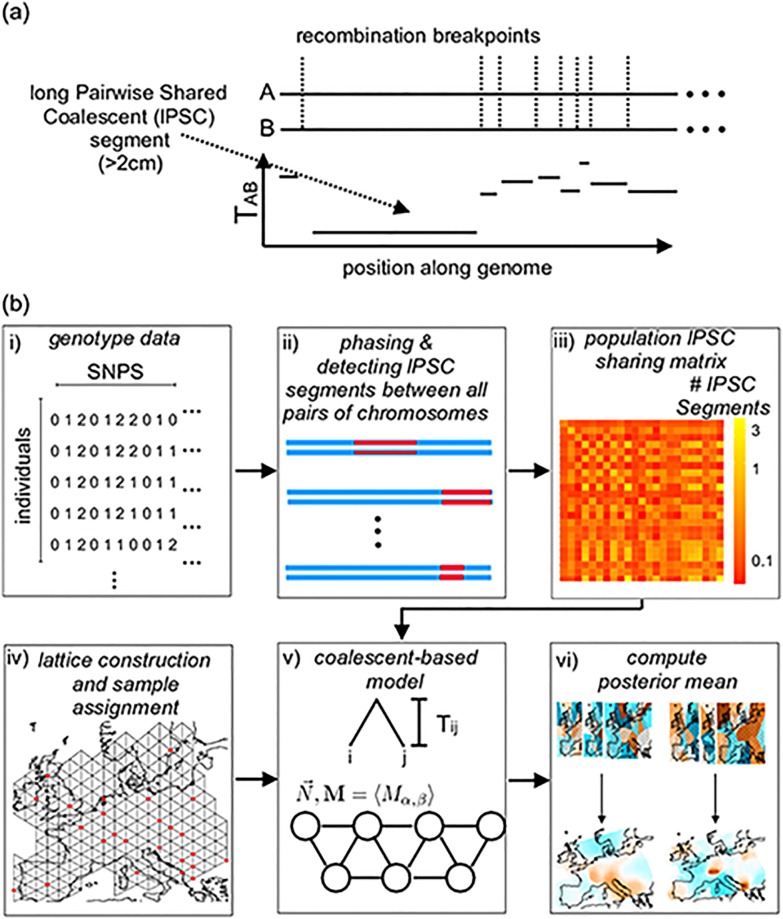
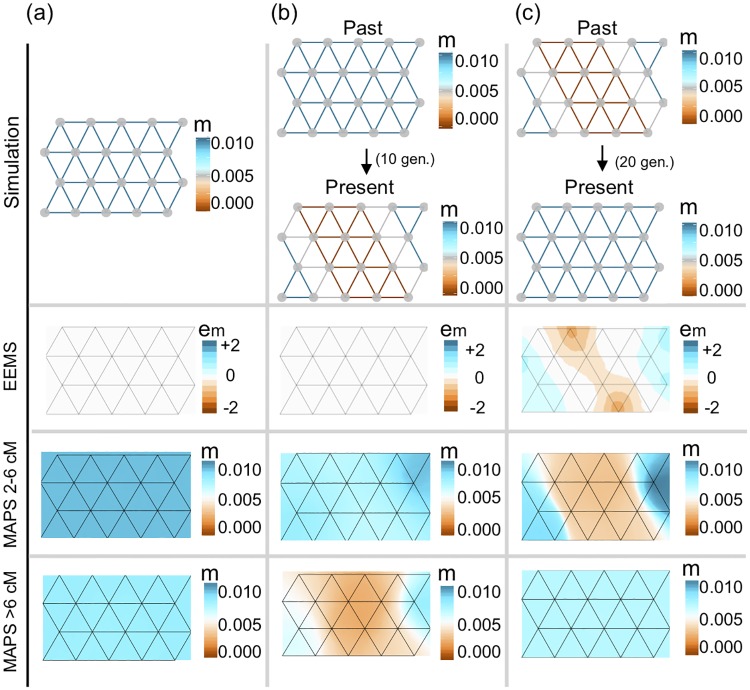
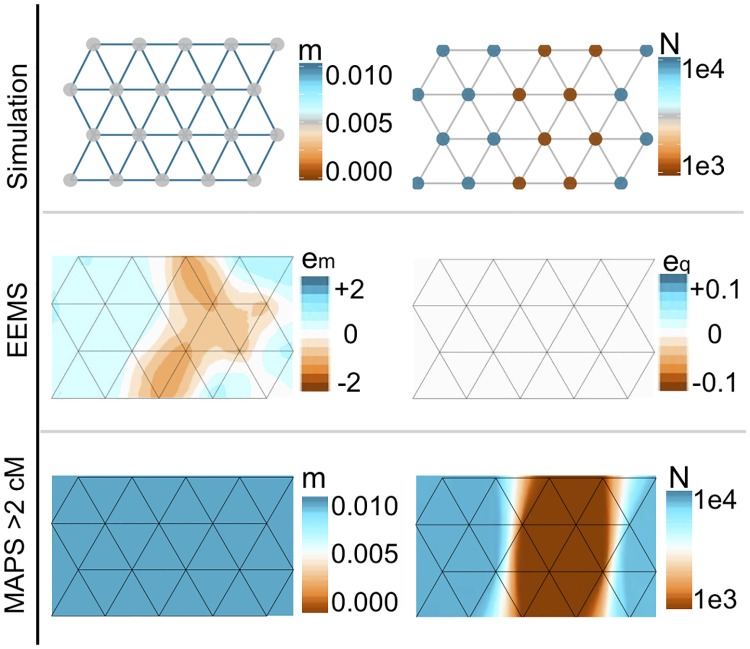
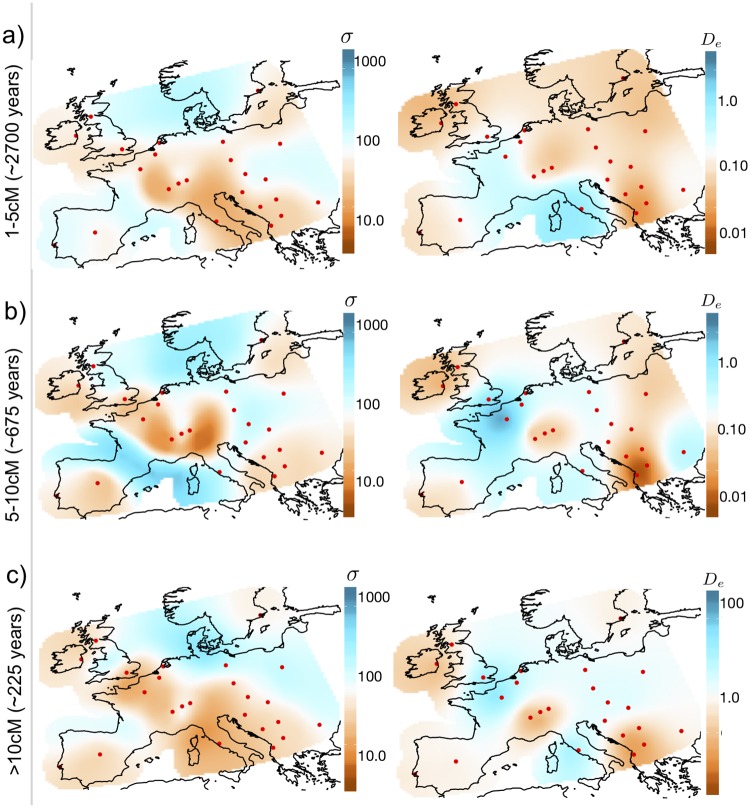
In many species a fundamental feature of genetic diversity is that genetic similarity decays with geographic distance; however, this relationship is often complex, and may vary across space and time. Methods to uncover and visualize such relationships have widespread use for analyses in molecular ecology, conservation genetics, evolutionary genetics, and human genetics. While several frameworks exist, a promising approach is to infer maps of how migration rates vary across geographic space. Such maps could, in principle, be estimated across time to reveal the full complexity of population histories. Here, we take a step in this direction: we present a method to infer maps of population sizes and migration rates associated with different time periods from a matrix of genetic similarity between every pair of individuals. Specifically, genetic similarity is measured by counting the number of long segments of haplotype sharing (also known as identity-by-descent tracts). By varying the length of these segments we obtain parameter estimates associated with different time periods. Using simulations, we show that the method can reveal time-varying migration rates and population sizes, including changes that are not detectable when using a similar method that ignores haplotypic structure. We apply the method to a dataset of contemporary European individuals (POPRES), and provide an integrated analysis of recent population structure and growth over the last ∼3,000 years in Europe.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Manel S, Schwartz MK, Luikart G, Taberlet P. Landscape Genetics: Combining Landscape Ecology and Population Genetics. Trends in Ecology & Evolution. 2003;18(4):189–197. 10.1016/S0169-5347(03)00008-9 - DOI
-
- Turner MG, Gardner RH, O’neill RV, et al. Landscape Ecology in Theory and Practice. Springer-Verlag; New York; 2001.
-
- Segelbacher G, Cushman SA, Epperson BK, Fortin MJ, Francois O, Hardy OJ, et al. Applications of Landscape Genetics in Conservation Biology: Concepts and Challenges. Conservation Genetics. 2010;11(2):375–385. 10.1007/s10592-009-0044-5 - DOI
-
- Rousset F. Genetic Structure and Selection in Subdivided Populations (MPB-40). Princeton University Press; New Jersey; 2004.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
