

Genomics of 1 million parent lifespans implicates novel pathways and common diseases and distinguishes survival chances
- PMID: 30642433
- PMCID: PMC6333444
- DOI: 10.7554/eLife.39856
Genomics of 1 million parent lifespans implicates novel pathways and common diseases and distinguishes survival chances
Abstract
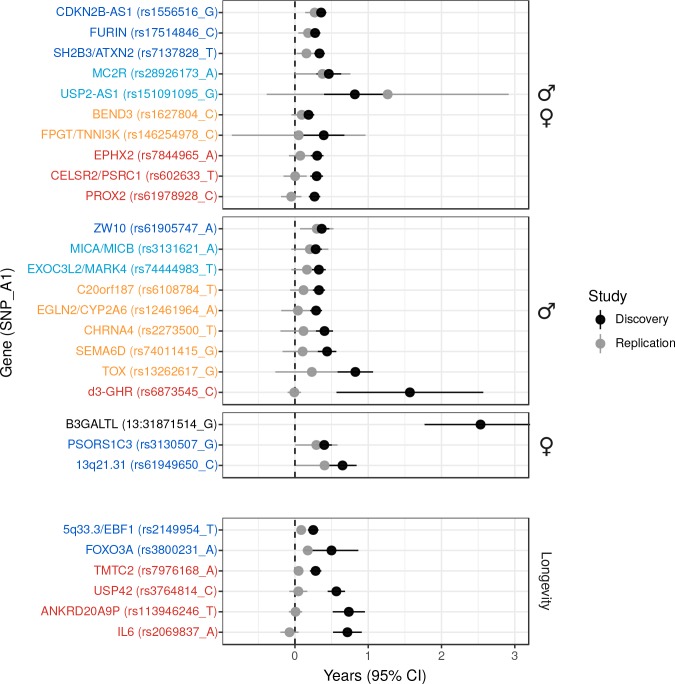
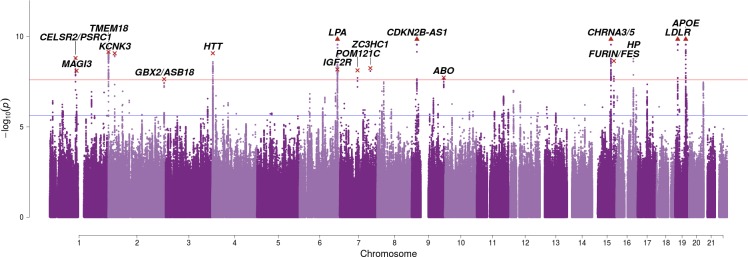
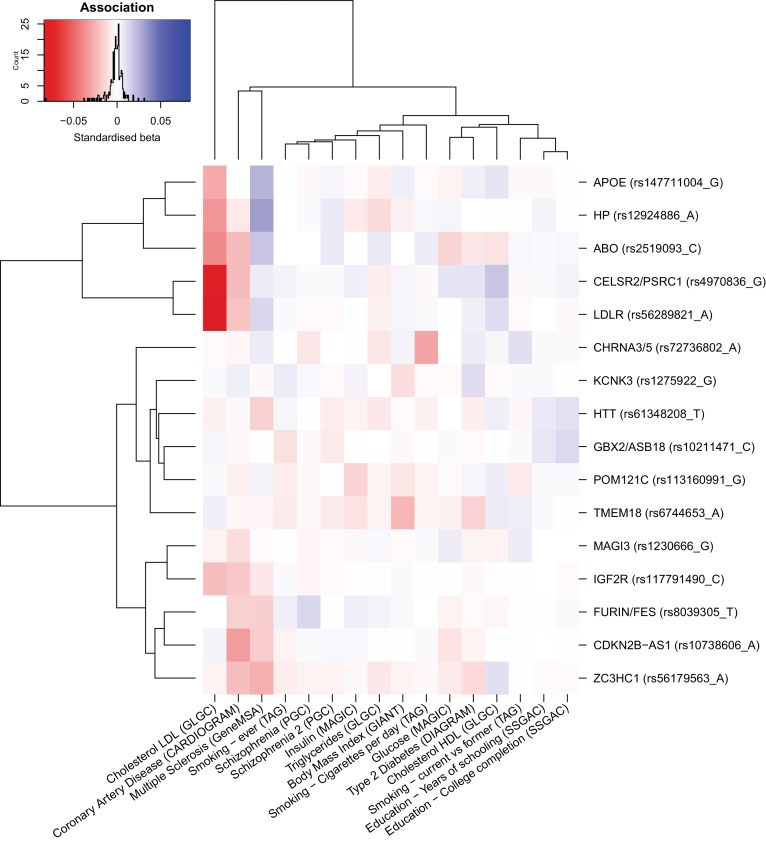
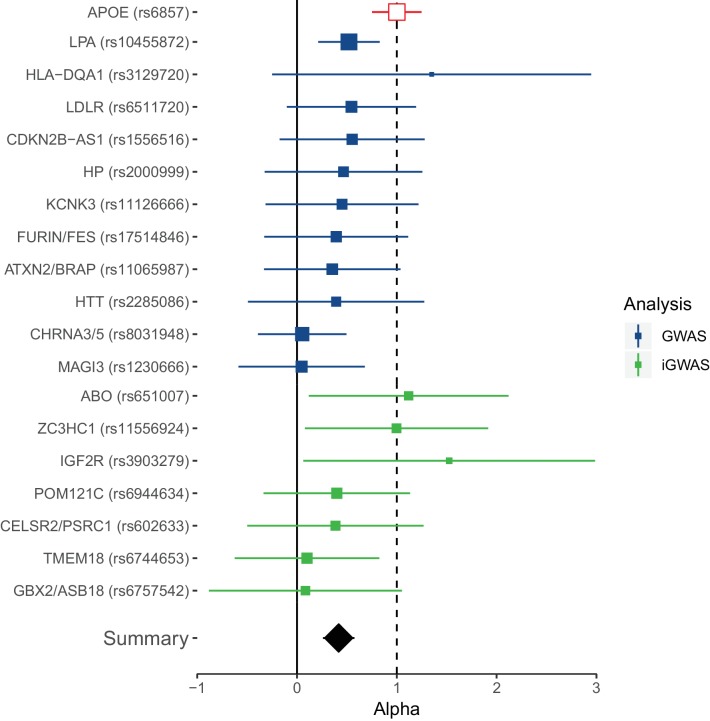
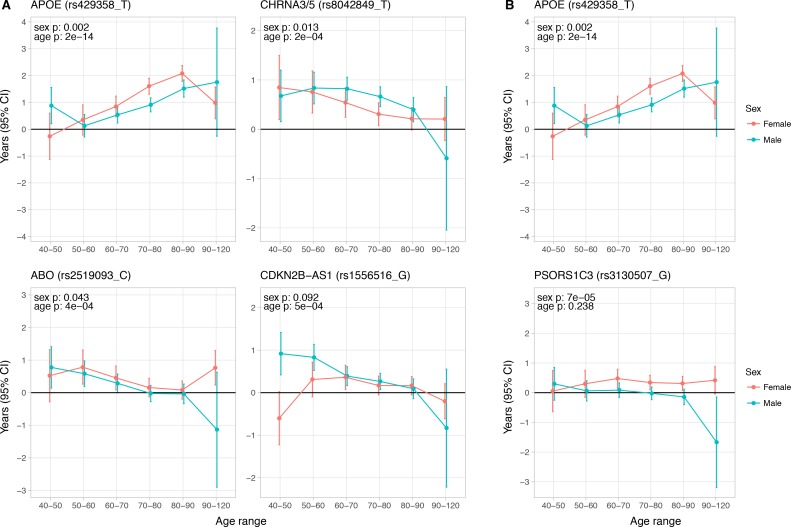
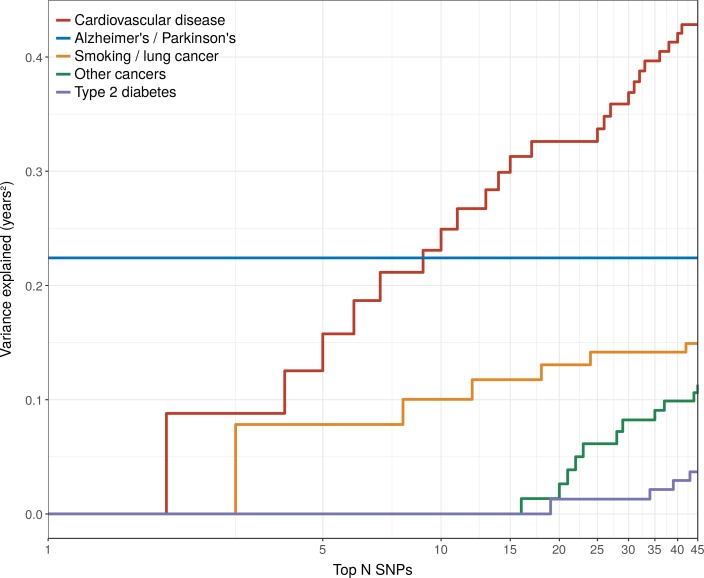
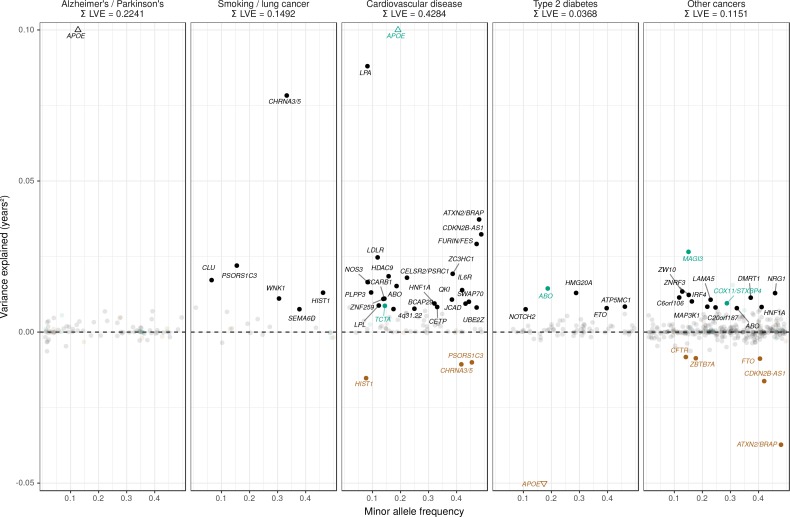
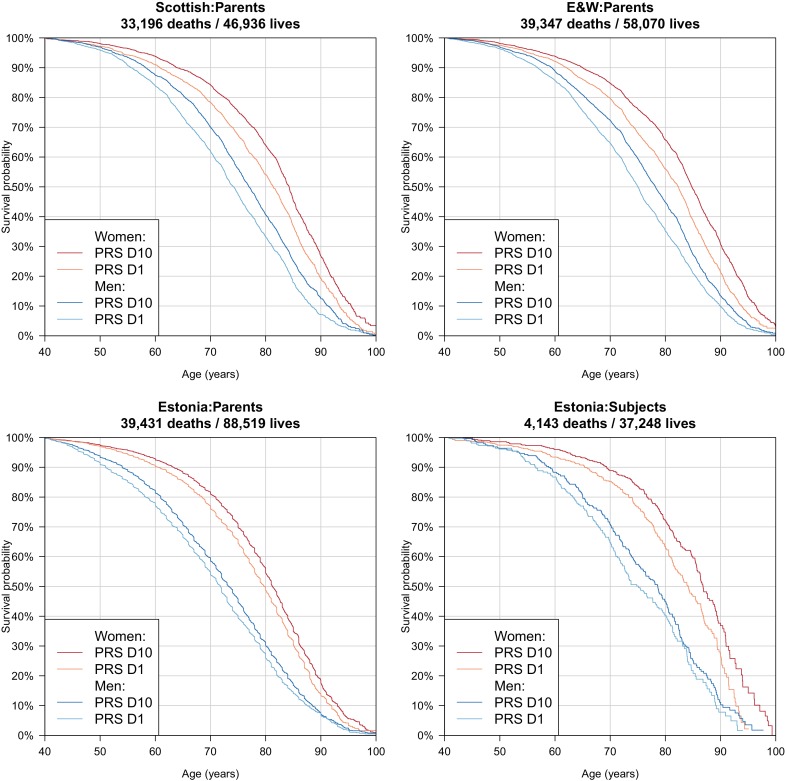
We use a genome-wide association of 1 million parental lifespans of genotyped subjects and data on mortality risk factors to validate previously unreplicated findings near CDKN2B-AS1, ATXN2/BRAP, FURIN/FES, ZW10, PSORS1C3, and 13q21.31, and identify and replicate novel findings near ABO, ZC3HC1, and IGF2R. We also validate previous findings near 5q33.3/EBF1 and FOXO3, whilst finding contradictory evidence at other loci. Gene set and cell-specific analyses show that expression in foetal brain cells and adult dorsolateral prefrontal cortex is enriched for lifespan variation, as are gene pathways involving lipid proteins and homeostasis, vesicle-mediated transport, and synaptic function. Individual genetic variants that increase dementia, cardiovascular disease, and lung cancer - but not other cancers - explain the most variance. Resulting polygenic scores show a mean lifespan difference of around five years of life across the deciles.
Editorial note: This article has been through an editorial process in which the authors decide how to respond to the issues raised during peer review. The Reviewing Editor's assessment is that all the issues have been addressed (see decision letter).
Keywords: complex trait; genetics; genomics; human; lifespan; longevity.
Plain language summary
Ageing happens to us all, and as the cabaret singer Maurice Chevalier pointed out, "old age is not that bad when you consider the alternative". Yet, the growing ageing population of most developed countries presents challenges to healthcare systems and government finances. For many older people, long periods of ill health are part of the end of life, and so a better understanding of ageing could offer the opportunity to prolong healthy living into old age. Ageing is complex and takes a long time to study – a lifetime in fact. This makes it difficult to discern its causes, among the countless possibilities based on an individual’s genes, behaviour or environment. While thousands of regions in an individual’s genetic makeup are known to influence their risk of different diseases, those that affect how long they will live have proved harder to disentangle. Timmers et al. sought to pinpoint such regions, and then use this information to predict, based on their DNA, whether someone had a better or worse chance of living longer than average. The DNA of over 500,000 people was read to reveal the specific ‘genetic fingerprints’ of each participant. Then, after asking each of the participants how long both of their parents had lived, Timmers et al. pinpointed 12 DNA regions that affect lifespan. Five of these regions were new and had not been linked to lifespan before. Across the twelve as a whole several were known to be involved in Alzheimer’s disease, smoking-related cancer or heart disease. Looking at the entire genome, Timmers et al. could then predict a lifespan score for each individual, and when they sorted participants into ten groups based on these scores they found that top group lived five years longer than the bottom, on average. Many factors beside genetics influence how long a person will live and our lifespan cannot be read from our DNA alone. Nevertheless, Timmers et al. had hoped to narrow down their search and discover specific genes that directly influence how quickly people age, beyond diseases. If such genes exist, their effects were too small to be detected in this study. The next step will be to expand the study to include more participants, which will hopefully pinpoint further genomic regions and help disentangle the biology of ageing and disease.
© 2019, Timmers et al.
Conflict of interest statement
PT, NM, KL, KF, ZN, XF, AB, DC, XS, ZK, JW, PJ No competing interests declared, TE Reviewing Editor, eLife
Figures


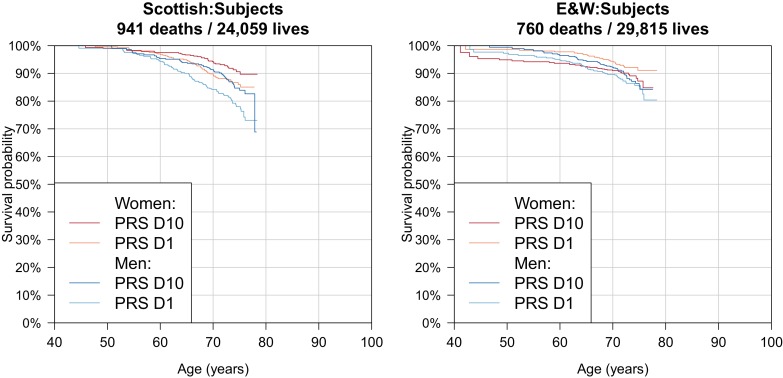



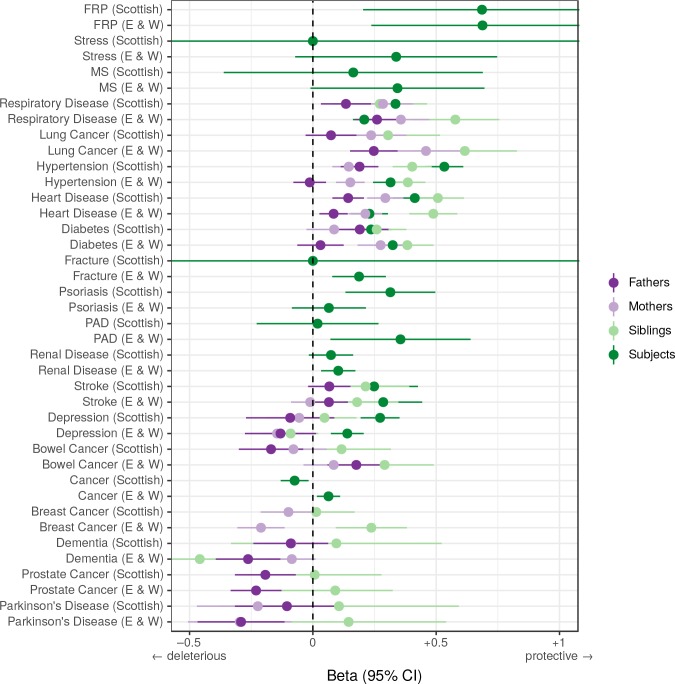
References
-
- Association of British Insurers and UK Government . Concordat and Moratorium on Genetics and Insurance. ABI; 2014.
-
- Battle A, Brown CD, Engelhardt BE, Montgomery SB, GTEx ConsortiumLaboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working GroupStatistical Methods groups—Analysis Working GroupEnhancing GTEx (eGTEx) groupsNIH Common FundNIH/NCINIH/NHGRINIH/NIMHNIH/NIDABiospecimen Collection Source Site—NDRIBiospecimen Collection Source Site—RPCIBiospecimen Core Resource—VARIBrain Bank Repository—University of Miami Brain Endowment BankLeidos Biomedical—Project ManagementELSI StudyGenome Browser Data Integration &Visualization—EBIGenome Browser Data Integration &Visualization—UCSC Genomics Institute, University of California Santa CruzLead analysts:Laboratory, Data Analysis &Coordinating Center (LDACC):NIH program management:Biospecimen collection:Pathology:eQTL manuscript working group: Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group. Statistical Methods groups—Analysis Working Group. Enhancing GTEx (eGTEx) groups. NIH Common Fund. NIH/NCI. NIH/NHGRI. NIH/NIMH. NIH/NIDA. Biospecimen Collection Source Site—NDRI. Biospecimen Collection Source Site—RPCI. Biospecimen Core Resource—VARI. Brain Bank Repository—University of Miami Brain Endowment Bank. Leidos Biomedical—Project Management. ELSI Study. Genome Browser Data Integration &Visualization—EBI. Genome Browser Data Integration &Visualization—UCSC Genomics Institute, University of California Santa Cruz. Lead analysts: Laboratory, Data Analysis &Coordinating Center (LDACC): NIH program management: Biospecimen collection: Pathology: eQTL manuscript working group: Genetic effects on gene expression across human tissues. Nature. 2017;550:204. doi: 10.1038/nature24277. - DOI - PMC - PubMed
-
- Beekman M, Blanché H, Perola M, Hervonen A, Bezrukov V, Sikora E, Flachsbart F, Christiansen L, De Craen AJ, Kirkwood TB, Rea IM, Poulain M, Robine JM, Valensin S, Stazi MA, Passarino G, Deiana L, Gonos ES, Paternoster L, Sørensen TI, Tan Q, Helmer Q, van den Akker EB, Deelen J, Martella F, Cordell HJ, Ayers KL, Vaupel JW, Törnwall O, Johnson TE, Schreiber S, Lathrop M, Skytthe A, Westendorp RG, Christensen K, Gampe J, Nebel A, Houwing-Duistermaat JJ, Slagboom PE, Franceschi C, GEHA consortium Genome-wide linkage analysis for human longevity: genetics of healthy aging study. Aging Cell. 2013;12:184–193. doi: 10.1111/acel.12039. - DOI - PMC - PubMed
-
- Ben-Avraham D, Govindaraju DR, Budagov T, Fradin D, Durda P, Liu B, Ott S, Gutman D, Sharvit L, Kaplan R, Bougnères P, Reiner A, Shuldiner AR, Cohen P, Barzilai N, Atzmon G. The GH receptor exon 3 deletion is a marker of male-specific exceptional longevity associated with increased GH sensitivity and taller stature. Science Advances. 2017;3:e1602025. doi: 10.1126/sciadv.1602025. - DOI - PMC - PubMed
-
- Broer L, Buchman AS, Deelen J, Evans DS, Faul JD, Lunetta KL, Sebastiani P, Smith JA, Smith AV, Tanaka T, Yu L, Arnold AM, Aspelund T, Benjamin EJ, De Jager PL, Eirkisdottir G, Evans DA, Garcia ME, Hofman A, Kaplan RC, Kardia SLR, Kiel DP, Oostra BA, Orwoll ES, Parimi N, Psaty BM, Rivadeneira F, Rotter JI, Seshadri S, Singleton A, Tiemeier H, Uitterlinden AG, Zhao W, Bandinelli S, Bennett DA, Ferrucci L, Gudnason V, Harris TB, Karasik D, Launer LJ, Perls TT, Slagboom PE, Tranah GJ, Weir DR, Newman AB, van Duijn CM, Murabito JM. GWAS of longevity in CHARGE consortium confirms APOE and FOXO3 candidacy. The Journals of Gerontology: Series A. 2015;70:110–118. doi: 10.1093/gerona/glu166. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- MC_UU_00007/10/MRC_/Medical Research Council/United Kingdom
- 204979/Z/16/Z/Edinburgh Clinical Academic Track/International
- 2014-00371/Svenska Forskningsrådet Formas/International
- MC_QA137853/MRC_/Medical Research Council/United Kingdom
- PUT 1665/Estonian Research Competency Council/International
- R01 HL105756/HL/NHLBI NIH HHS/United States
- DTP in Precision Medicine MR/N013166/1,HGU QTL in health and disease/MRC_/Medical Research Council/United Kingdom
- 208806/Z/17/Z/WT_/Wellcome Trust/United Kingdom
- MC_PC_17228/MRC_/Medical Research Council/United Kingdom
- 31003A_169929/Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung/International
- R01 DK075787/DK/NIDDK NIH HHS/United States
- 2017-02543/Svenska Forskningsrådet Formas/International
- PhD Training Fellowship for Clinicians/WT_/Wellcome Trust/United Kingdom
- 51RTP0_151019/SystemsX.ch/International
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
