

Integration of Genetic Testing and Pathology for the Diagnosis of Adults with FSGS
- PMID: 30647093
- PMCID: PMC6390925
- DOI: 10.2215/CJN.08750718
Integration of Genetic Testing and Pathology for the Diagnosis of Adults with FSGS
Abstract
Background and objectives: FSGS and nephrotic syndrome studies have shown that single gene causes are more likely to be found in pediatric cases than adults. Consequently, many studies have examined limited gene panels in largely pediatric cohorts.
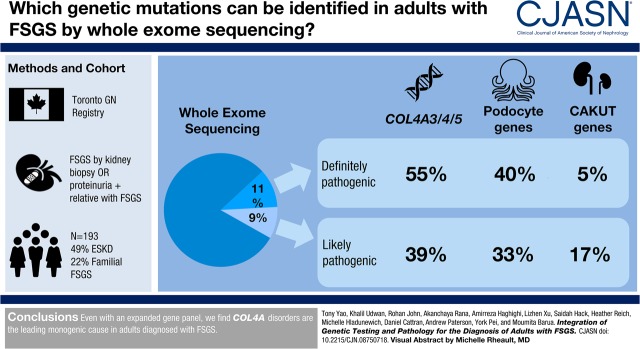
Design, setting, participants, & measurements: Whole-exome sequencing was performed in adults with FSGS diagnosed between 1976 and 2017 in the Toronto GN Registry. An expanded panel of 109 genes linked to FSGS, glomerular basement membrane abnormalities, as well as causes of pediatric ESKD including congenital abnormalities of the kidney and urinary tract (CAKUT) and nephronophthisis, were examined.
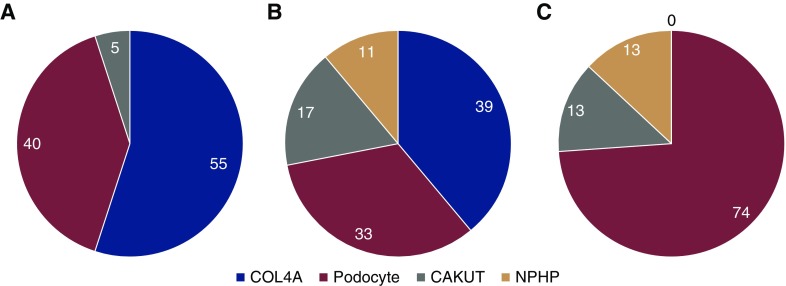
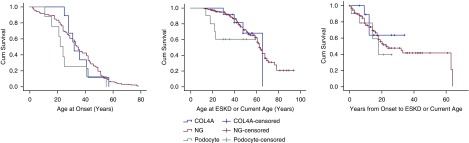
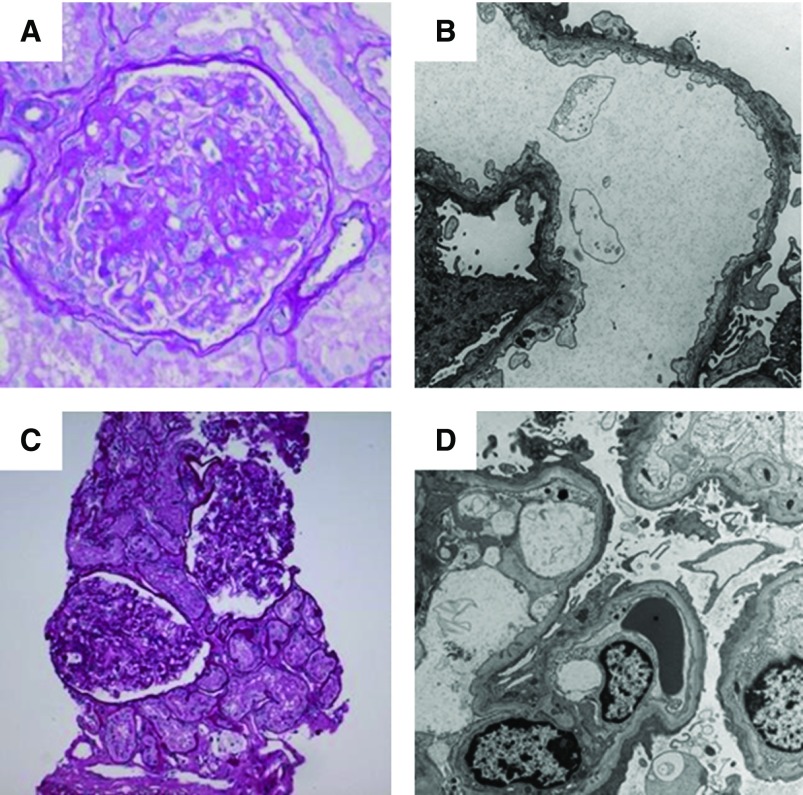
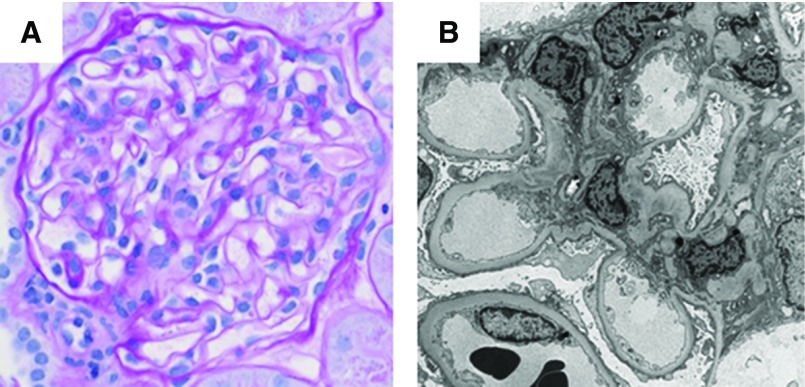
Results: The cohort was composed of 193 individuals from 179 families. Nearly half (49%) developed ESKD at a mean age of 47±17 years. The genetic diagnostic rate was 11%. Of definitely pathogenic variants, 55% were in COL4A (A3/A4/A5), 40% were in podocyte genes, and 5% were in CAKUT genes. Many, but not all individuals with COL4A definitely pathogenic variants had some evidence of glomerular basement membrane abnormalities. The estimated mean survival/age of kidney failure for individuals with COL4A definitely pathogenic variants was 58 years (95% confidence interval, 49 to 69), far later than what has been reported in the literature. Likely pathogenic variants were identified in an additional 9% of the cohort, with most in COL4A. Correlation with glomerular basement membrane morphology suggested a causal role for at least some of these likely pathogenic variants.
Conclusions: Even with an expanded gene panel, we find that COL4A disorders are the leading monogenic cause in adults diagnosed with FSGS.
Podcast: This article contains a podcast at https://www.asn-online.org/media/podcast/CJASN/2019_01_15_CJASNPodcast_19_02_.mp3.
Keywords: Cohort Studies; FSGS; Genetic Testing; Glomerular Basement Membrane; Kidney Failure, Chronic; Podocytes; Registries; Renal Insufficiency; Urogenital Abnormalities; Whole Exome Sequencing; glomerulonephritis; human genetics; idiopathic nephrotic syndrome; kidney; nephrotic syndrome; renal development; type 4A collagen; vesico-ureteral reflux.
Copyright © 2019 by the American Society of Nephrology.
Figures
References
-
- Falk R: JC, Nachman P: Primary glomerular disease. In: The Kidney, edited by Brenner BM, Philadelphia, W. B. Saunders Company, 2000, pp 1263–1332
-
- Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, Mathis BJ, Rodríguez-Pérez JC, Allen PG, Beggs AH, Pollak MR: Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 24: 251–256, 2000 - PubMed
-
- Reiser J, Polu KR, Möller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR: TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet 37: 739–744, 2005 - PMC - PubMed
-
- Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB: A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 308: 1801–1804, 2005 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
