

Selenoprotein M Promotes Hypothalamic Leptin Signaling and Thioredoxin Antioxidant Activity
- PMID: 30648404
- PMCID: PMC8617589
- DOI: 10.1089/ars.2018.7594
Selenoprotein M Promotes Hypothalamic Leptin Signaling and Thioredoxin Antioxidant Activity
Abstract
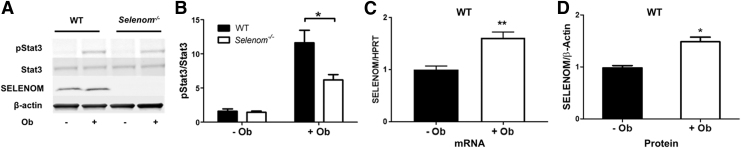
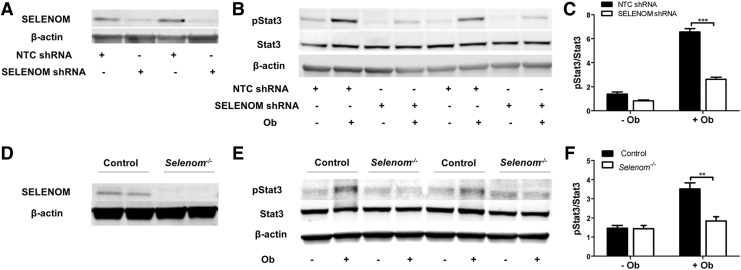
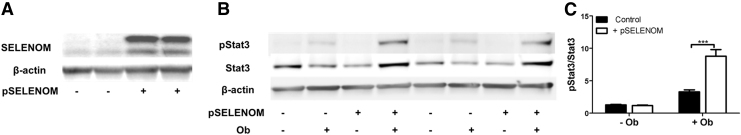
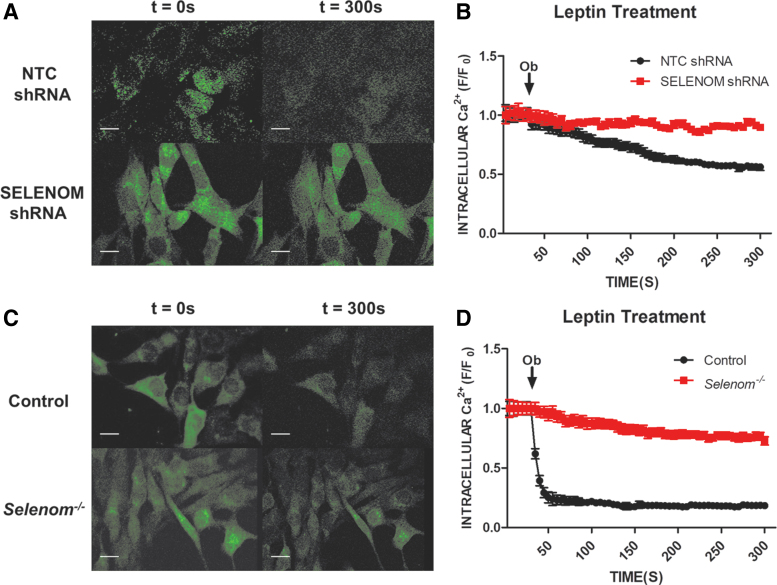
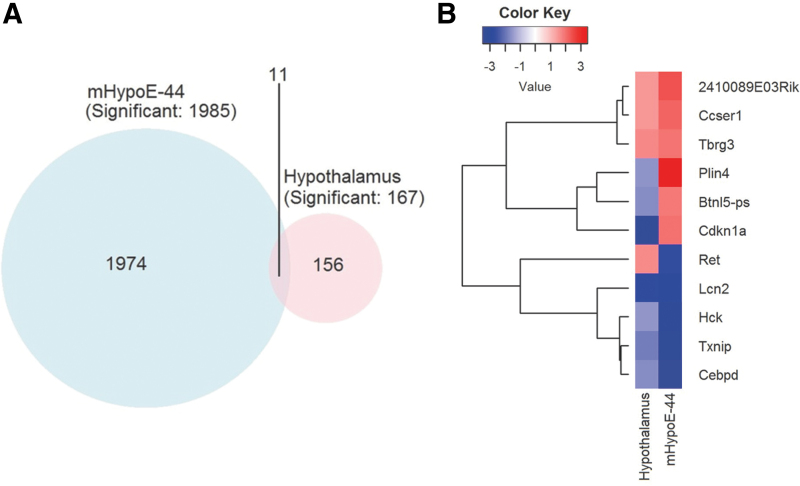
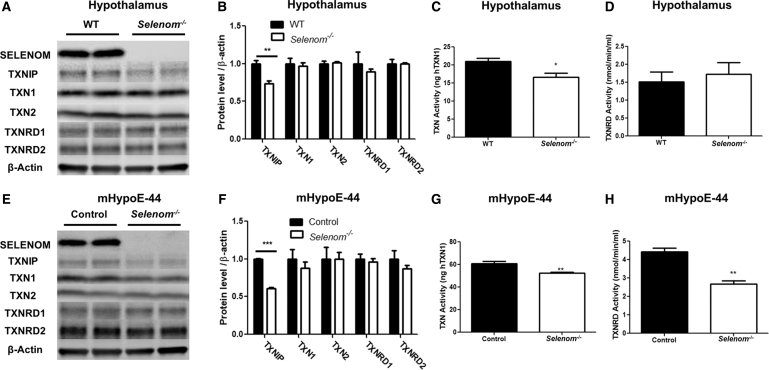
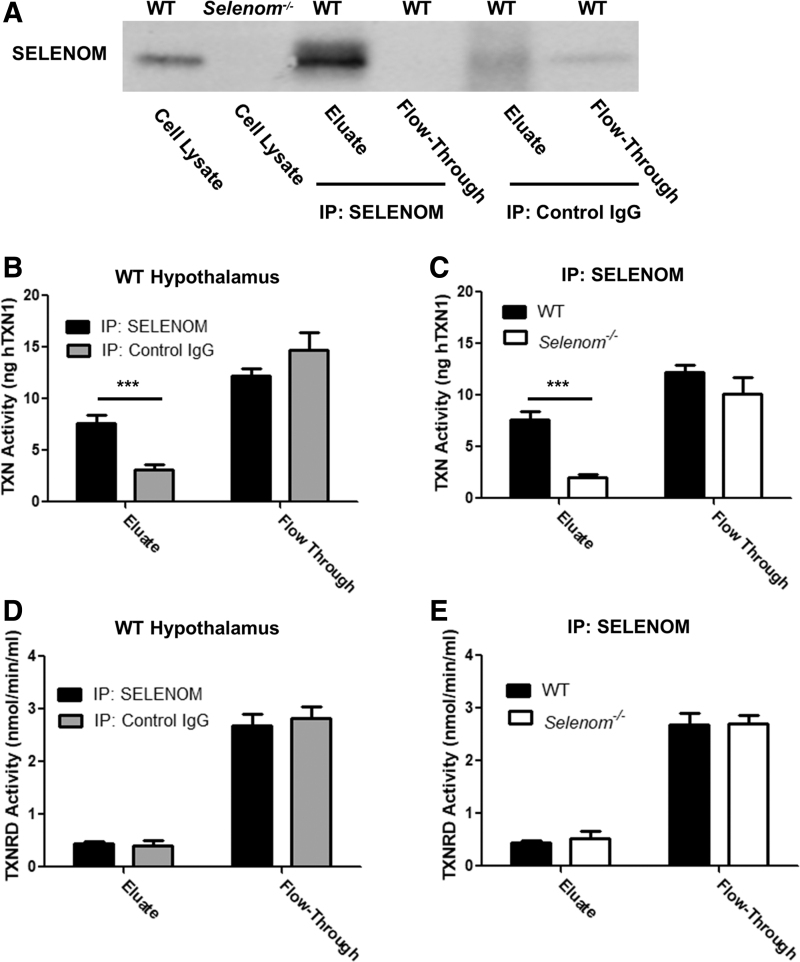
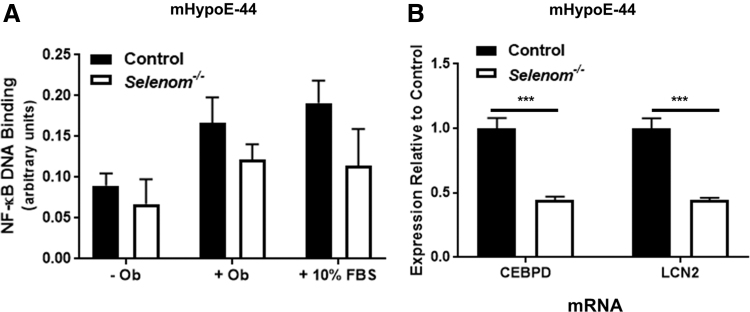
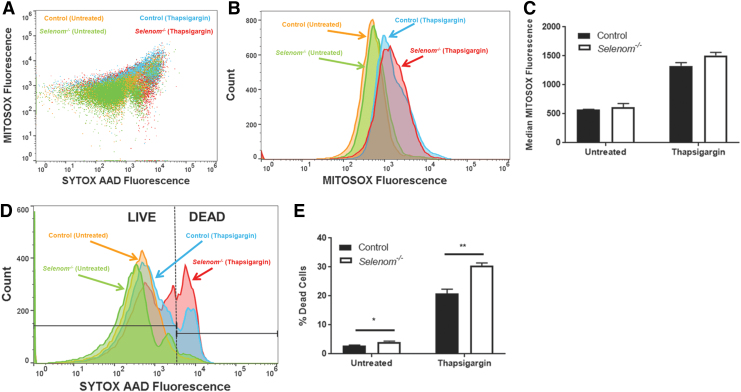
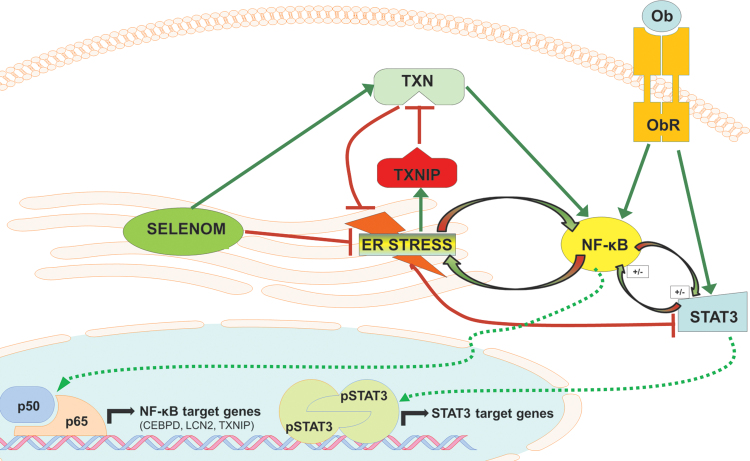
Aims: Selenoproteins are an essential class of proteins involved in redox signaling and energy metabolism. However, the functions of many selenoproteins are not clearly established. Selenoprotein M (SELENOM), an endoplasmic reticulum (ER)-resident oxidoreductase bearing structural similarity to thioredoxin (TXN), is among those yet to be fully characterized. This protein is highly expressed in hypothalamic regions involved in leptin signaling and has been previously linked to energy metabolism. Herein, we performed a series of studies using in vivo and in vitro models to probe the specific influence of SELENOM on hypothalamic leptin signaling and assess SELENOM-regulated pathways. Innovation and Results: Our initial experiment in vivo demonstrated that (i) leptin promotes hypothalamic expression of SELENOM and (ii) leptin-induced STAT3 phosphorylation is impeded by SELENOM deficiency. Additional in vitro studies using mHypoE-44 immortalized hypothalamic neurons corroborated these findings, as SELENOM deficiency obstructed downstream STAT3 phosphorylation and cytosolic calcium responses evoked by leptin treatment. Correspondingly, SELENOM overexpression enhanced leptin sensitivity. Microarray analysis conducted in parallel on hypothalamic tissue and mHypoE-44 cells revealed multiple genes significantly affected by SELENOM deficiency, including thioredoxin interacting protein, a negative regulator of the TXN system. Further analysis determined that (i) SELENOM itself possesses intrinsic TXN activity and (ii) SELENOM deficiency leads to a reduction in overall TXN activity. Finally, mHypoE-44 cells lacking SELENOM displayed diminished activation of the nuclear factor kappa-light-chain enhancer of activated B-cells (NF-κB) signaling pathway and increased susceptibility to ER stress-mediated cell death. Conclusion: In sum, these findings establish SELENOM as a positive regulator of leptin signaling and TXN antioxidant activity in the hypothalamus. Antioxid. Redox Signal. 35, 775-787.
Keywords: endoplasmic reticulum; hypothalamus; leptin; selenoprotein; thioredoxin.
Conflict of interest statement
No competing financial interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
