

Broad range of missense error frequencies in cellular proteins
- PMID: 30649420
- PMCID: PMC6451103
- DOI: 10.1093/nar/gky1319
Broad range of missense error frequencies in cellular proteins
Abstract
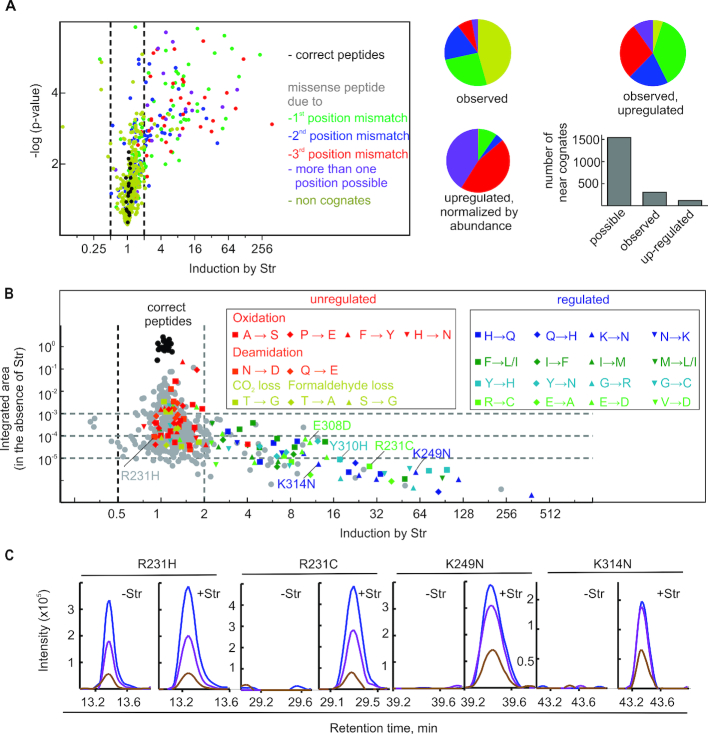
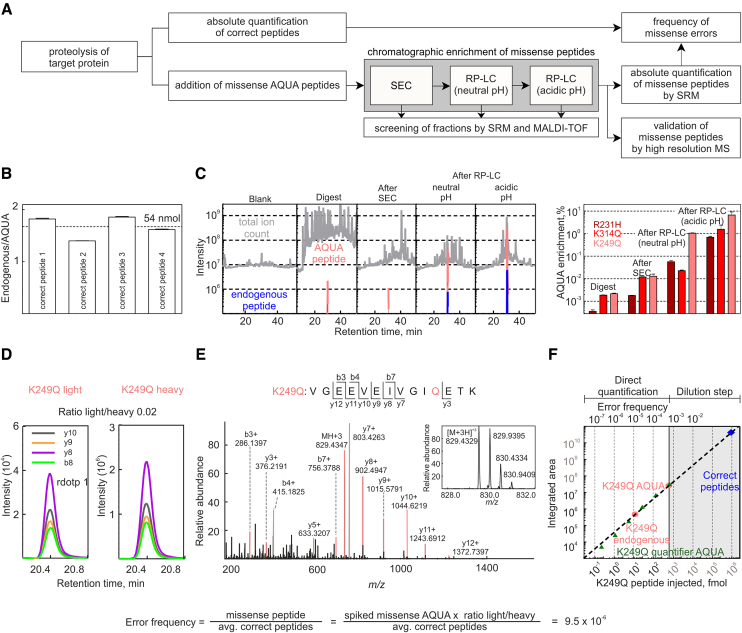
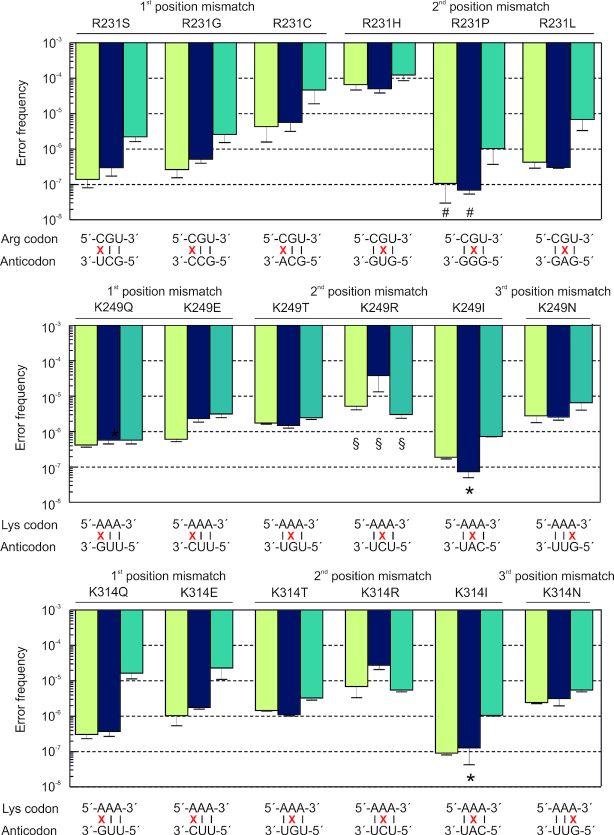
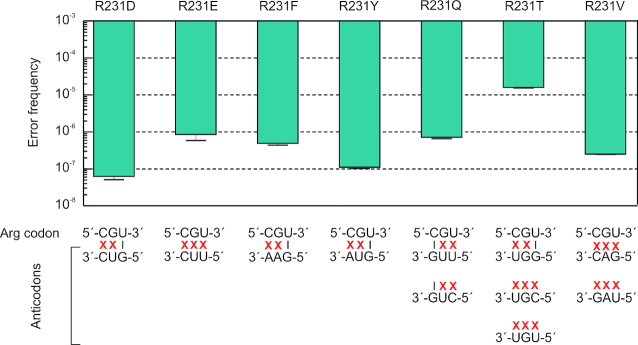
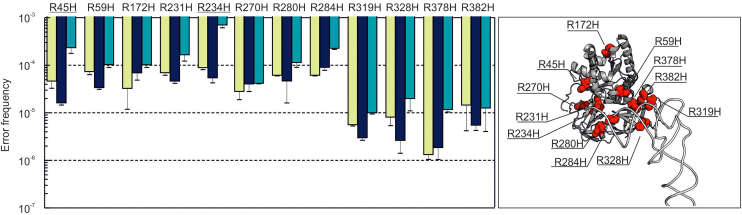
Assessment of the fidelity of gene expression is crucial to understand cell homeostasis. Here we present a highly sensitive method for the systematic Quantification of Rare Amino acid Substitutions (QRAS) using absolute quantification by targeted mass spectrometry after chromatographic enrichment of peptides with missense amino acid substitutions. By analyzing incorporation of near- and non-cognate amino acids in a model protein EF-Tu, we show that most of missense errors are too rare to detect by conventional methods, such as DDA, and are estimated to be between <10-7-10-5 by QRAS. We also observe error hotspots of up to 10-3 for some types of mismatches, including the G-U mismatch. The error frequency depends on the expression level of EF-Tu and, surprisingly, the amino acid position in the protein. QRAS is not restricted to any particular miscoding event, organism, strain or model protein and is a reliable tool to analyze very rare proteogenomic events.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Frederico L.A., Kunkel T.A., Shaw B.R.. A sensitive genetic assay for the detection of cytosine deamination: Determination of rate constants and the activation energy. Biochemistry. 1990; 29:2532–2537. - PubMed
-
- Pakula A.A., Sauer R.T.. Genetic analysis of protein stability and function. Annu. Rev. Genet. 1989; 23:289–310. - PubMed
-
- Tokuriki N., Tawfik D.S.. Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature. 2009; 459:668–673. - PubMed
-
- Stefani M., Dobson C.M.. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J Mol. Med. (Berl.). 2003; 81:678–699. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
