

Fine-mapping cis-regulatory variants in diverse human populations
- PMID: 30650056
- PMCID: PMC6335058
- DOI: 10.7554/eLife.39595
Fine-mapping cis-regulatory variants in diverse human populations
Abstract
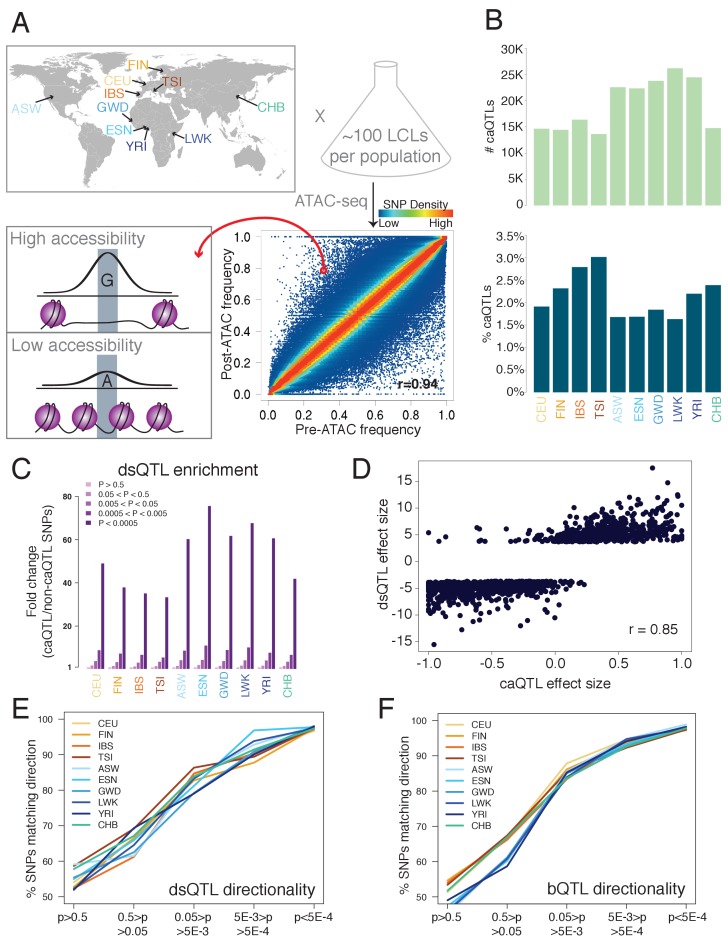
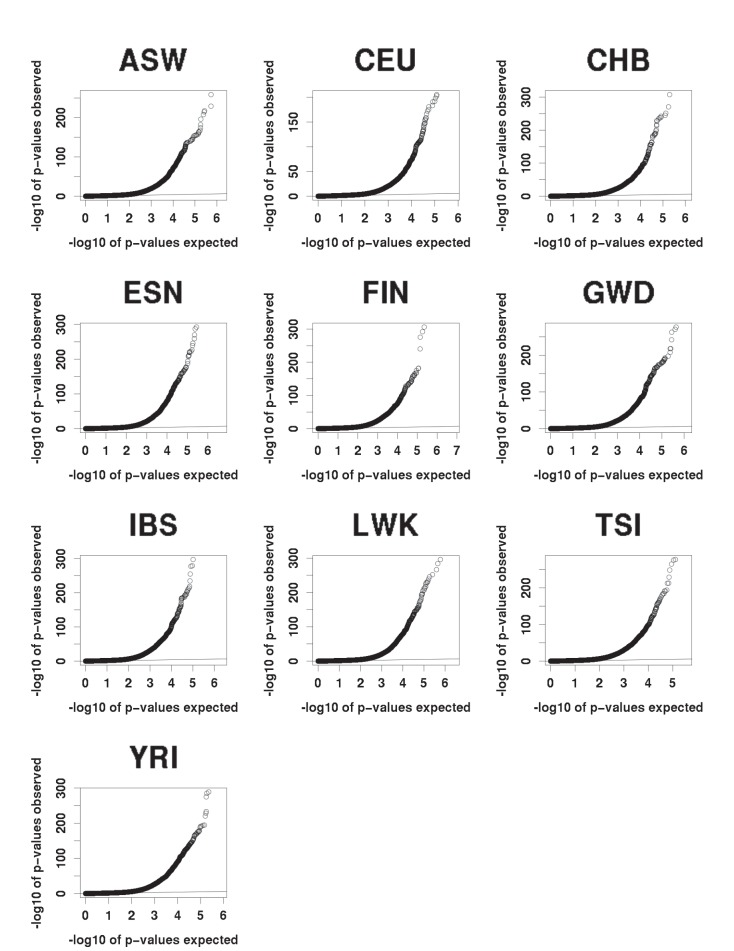
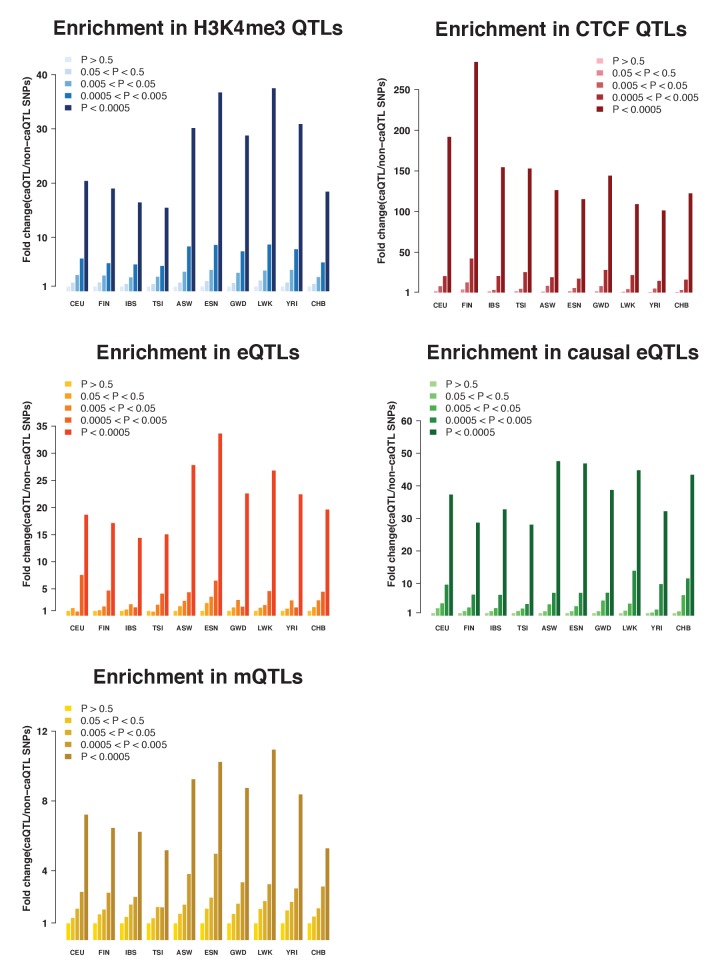
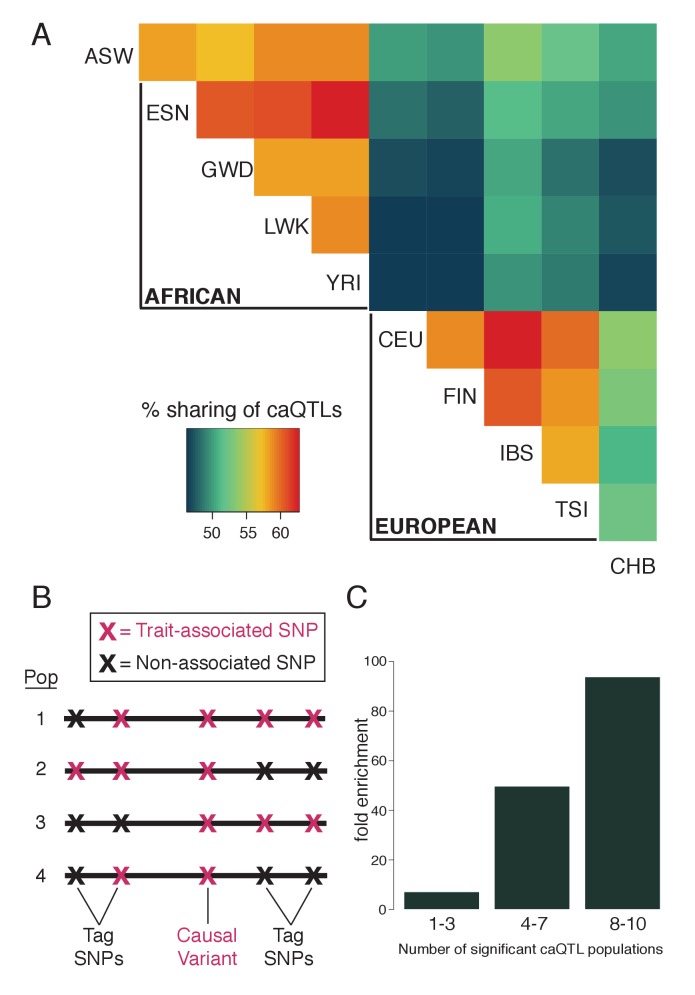
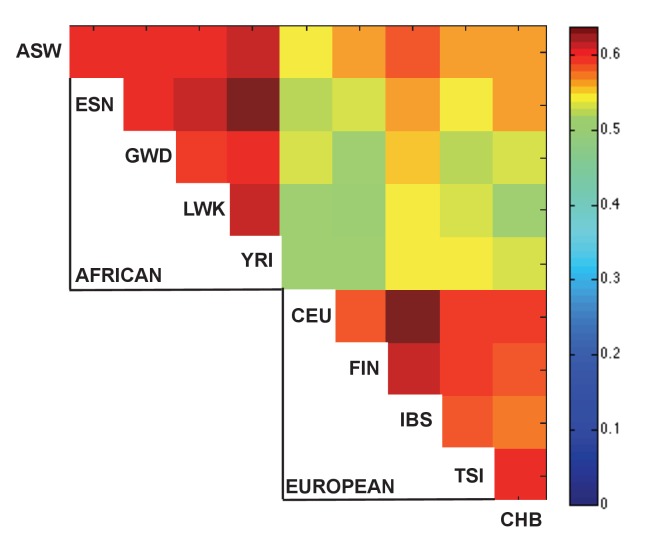
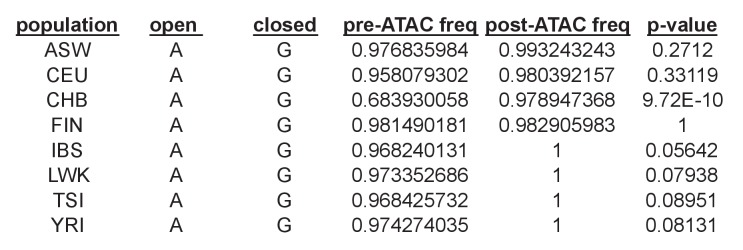
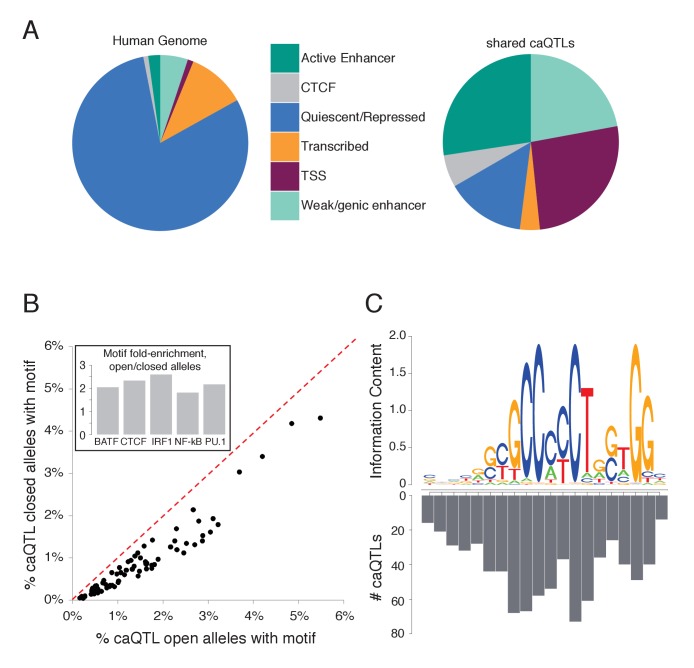
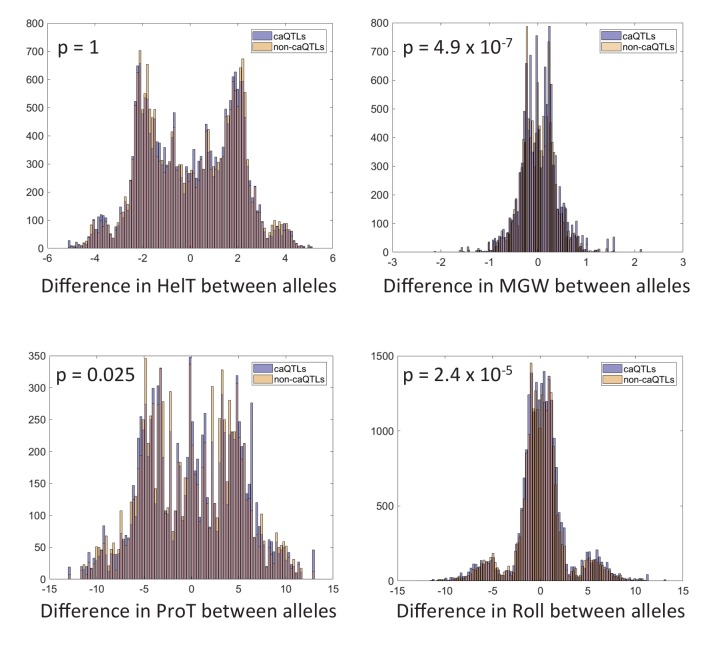
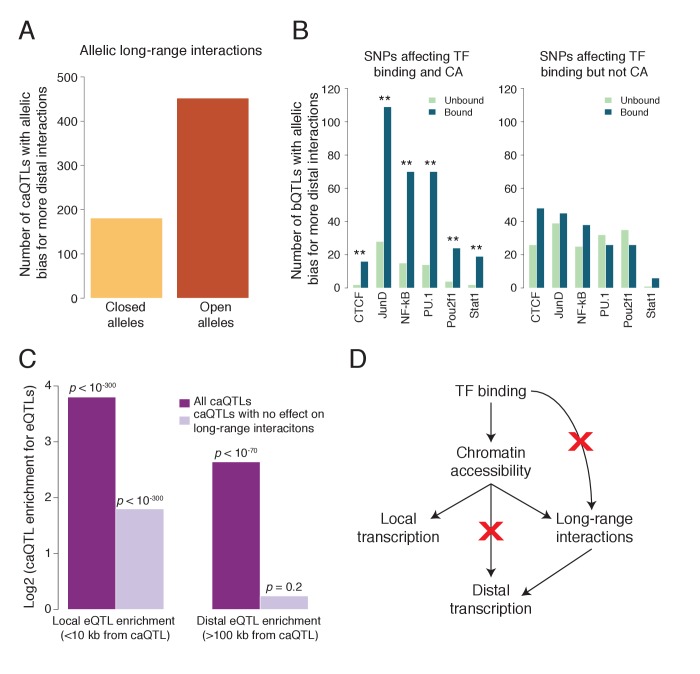
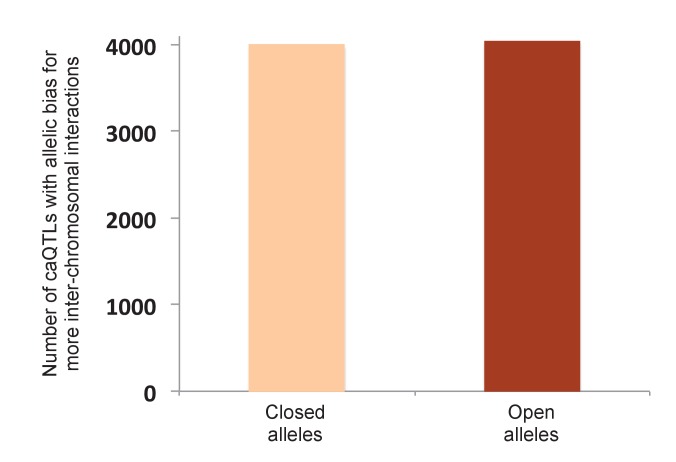
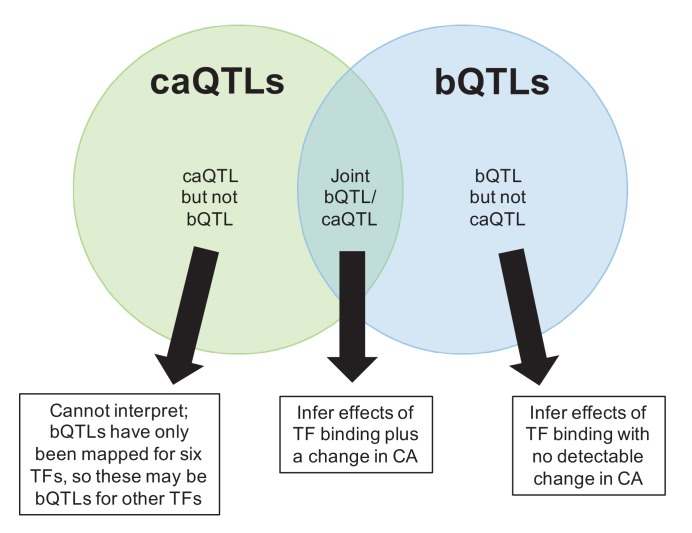
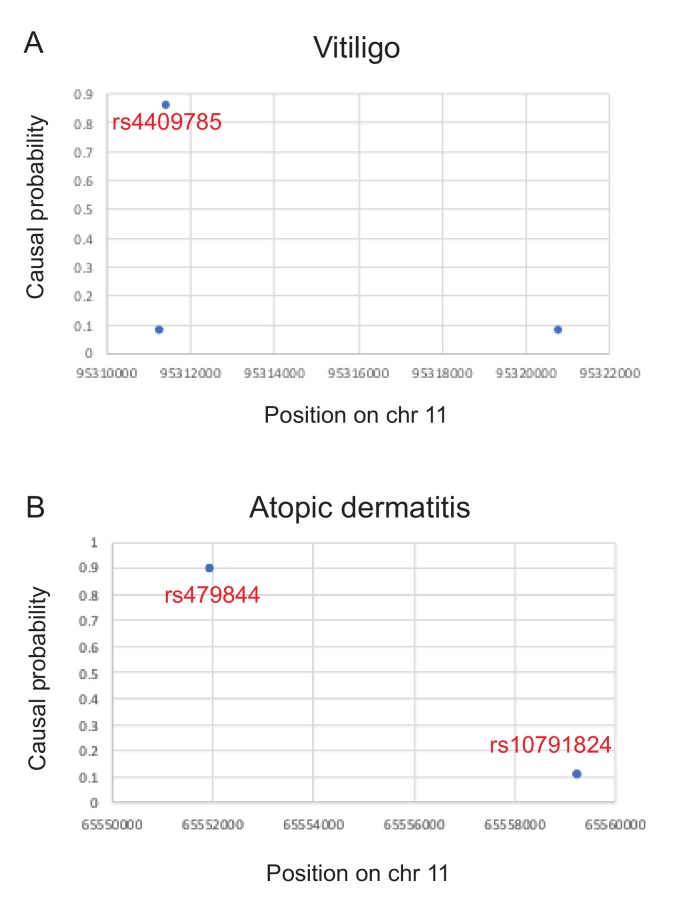
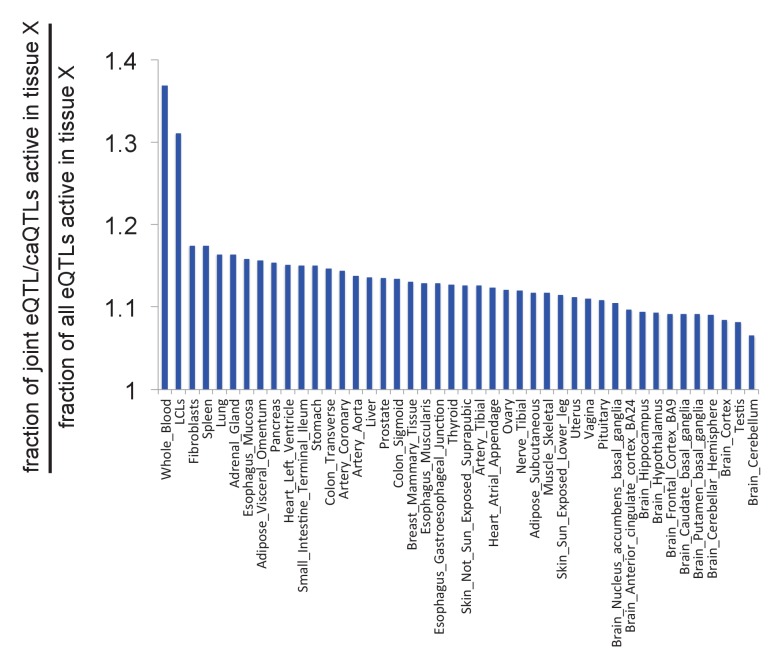
Genome-wide association studies (GWAS) are a powerful approach for connecting genotype to phenotype. Most GWAS hits are located in cis-regulatory regions, but the underlying causal variants and their molecular mechanisms remain unknown. To better understand human cis-regulatory variation, we mapped quantitative trait loci for chromatin accessibility (caQTLs)-a key step in cis-regulation-in 1000 individuals from 10 diverse populations. Most caQTLs were shared across populations, allowing us to leverage the genetic diversity to fine-map candidate causal regulatory variants, several thousand of which have been previously implicated in GWAS. In addition, many caQTLs that affect the expression of distal genes also alter the landscape of long-range chromosomal interactions, suggesting a mechanism for long-range expression QTLs. In sum, our results show that molecular QTL mapping integrated across diverse populations provides a high-resolution view of how worldwide human genetic variation affects chromatin accessibility, gene expression, and phenotype.
Editorial note: This article has been through an editorial process in which the authors decide how to respond to the issues raised during peer review. The Reviewing Editor's assessment is that minor issues remain unresolved (see decision letter).
Keywords: chromatin; chromosomes; fine-mapping; gene expression; genetics; genomics; gwas; human; qtl; transcription.
© 2019, Tehranchi et al.
Conflict of interest statement
AT, BH, MD, IK, KP, PC, HF No competing interests declared
Figures
References
-
- Banovich NE, Lan X, McVicker G, van de Geijn B, Degner JF, Blischak JD, Roux J, Pritchard JK, Gilad Y. Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLOS Genetics. 2014;10:e1004663. doi: 10.1371/journal.pgen.1004663. - DOI - PMC - PubMed
-
- Battle A, Brown CD, Engelhardt BE, Montgomery SB, GTEx Consortium. Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group. Statistical Methods groups—Analysis Working Group. Enhancing GTEx (eGTEx) groups. NIH Common Fund. NIH/NCI. NIH/NHGRI. NIH/NIMH. NIH/NIDA. Biospecimen Collection Source Site—NDRI. Biospecimen Collection Source Site—RPCI. Biospecimen Core Resource—VARI. Brain Bank Repository—University of Miami Brain Endowment Bank. Leidos Biomedical—Project Management. ELSI Study. Genome Browser Data Integration &Visualization—EBI. Genome Browser Data Integration &Visualization—UCSC Genomics Institute, University of California Santa Cruz. Lead analysts: Laboratory, Data Analysis &Coordinating Center (LDACC) NIH program management. Biospecimen collection. Pathology: eQTL manuscript working group Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
