

Neuroinflammation, Microglia, and Cell-Association during Prion Disease
- PMID: 30650564
- PMCID: PMC6356204
- DOI: 10.3390/v11010065
Neuroinflammation, Microglia, and Cell-Association during Prion Disease
Abstract
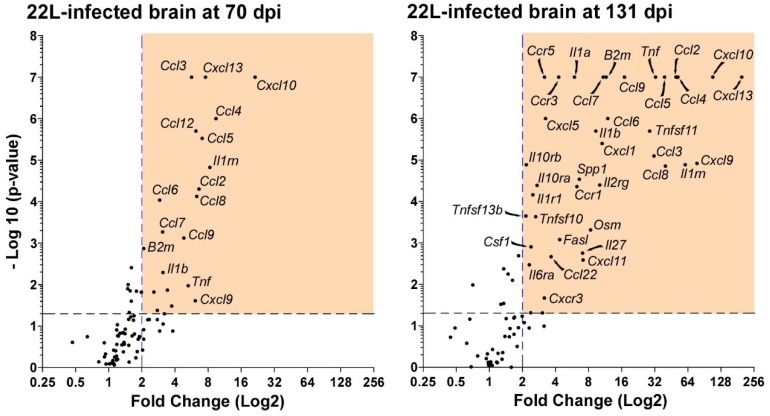
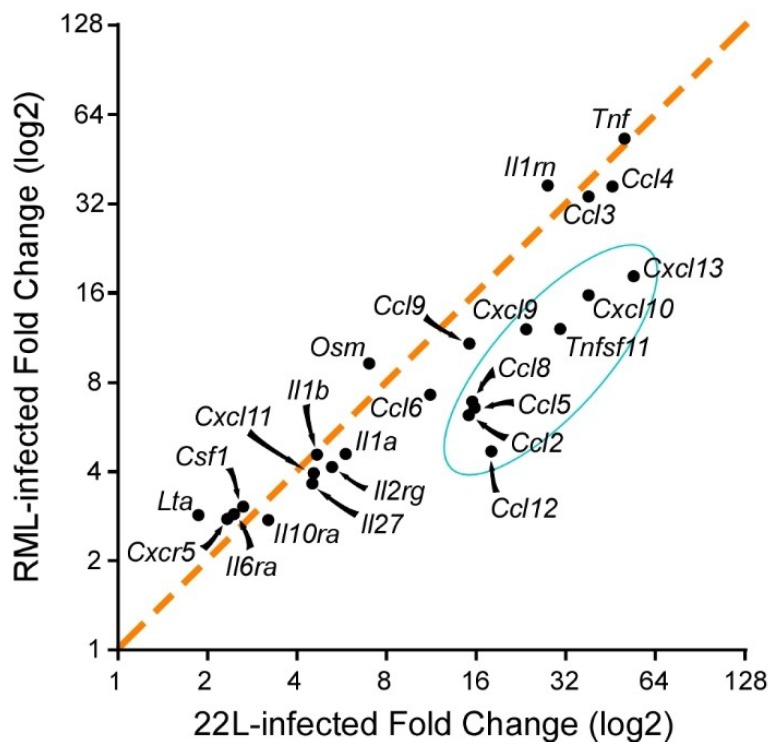
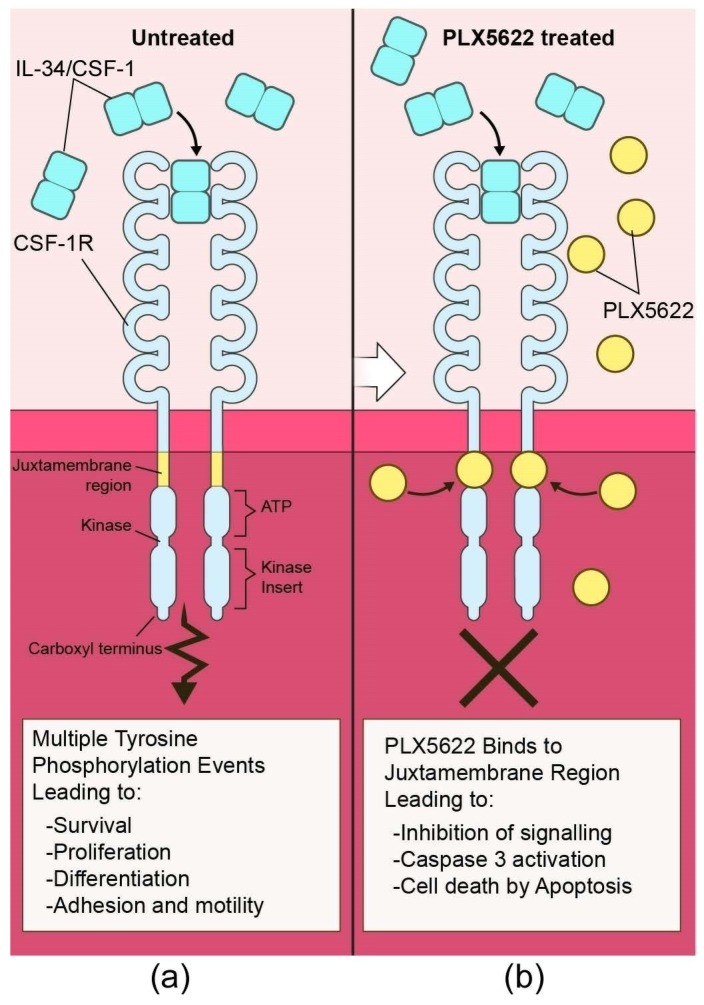
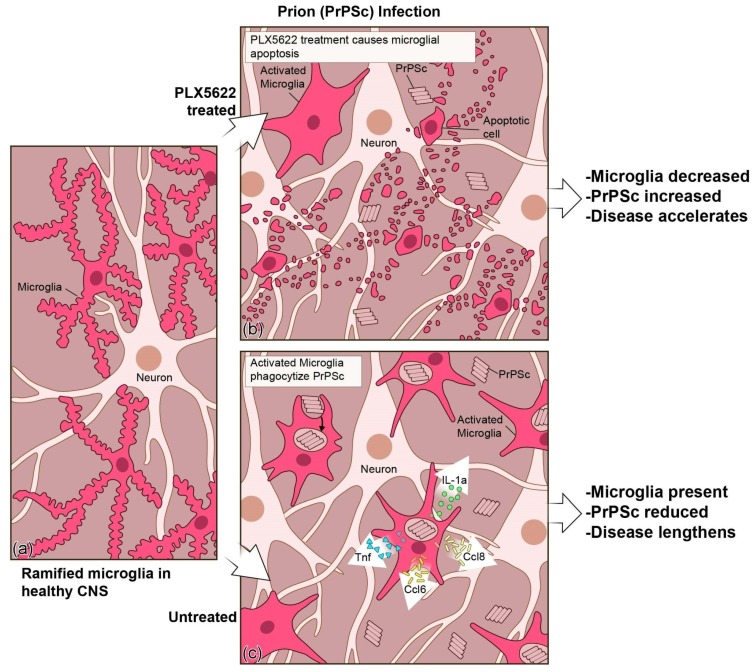
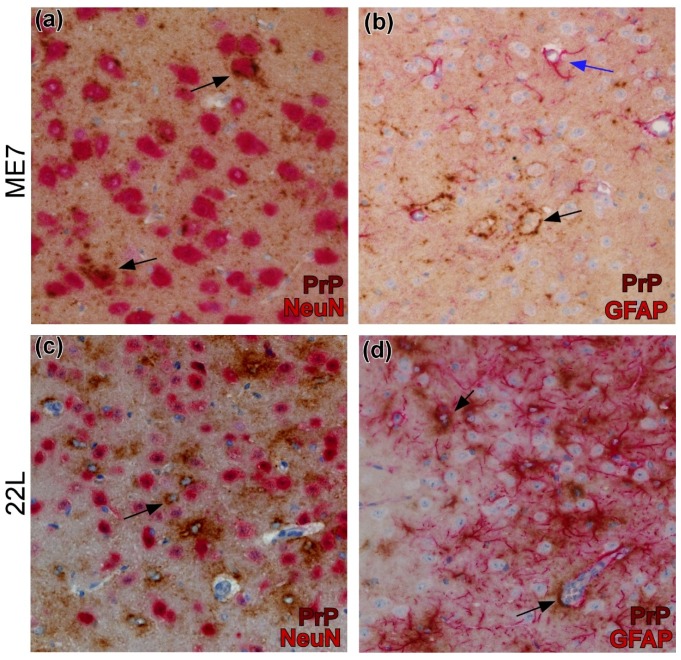
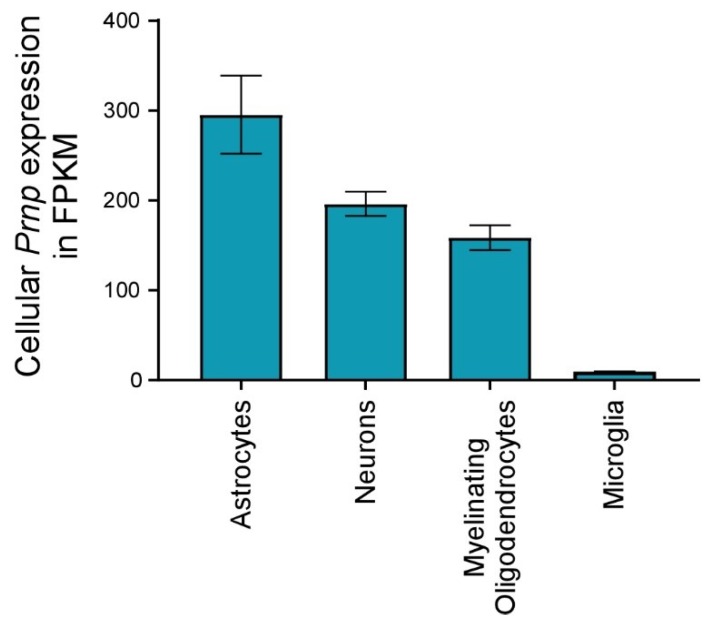
Prion disorders are transmissible diseases caused by a proteinaceous infectious agent that can infect the lymphatic and nervous systems. The clinical features of prion diseases can vary, but common hallmarks in the central nervous system (CNS) are deposition of abnormally folded protease-resistant prion protein (PrPres or PrPSc), astrogliosis, microgliosis, and neurodegeneration. Numerous proinflammatory effectors expressed by astrocytes and microglia are increased in the brain during prion infection, with many of them potentially damaging to neurons when chronically upregulated. Microglia are important first responders to foreign agents and damaged cells in the CNS, but these immune-like cells also serve many essential functions in the healthy CNS. Our current understanding is that microglia are beneficial during prion infection and critical to host defense against prion disease. Studies indicate that reduction of the microglial population accelerates disease and increases PrPSc burden in the CNS. Thus, microglia are unlikely to be a foci of prion propagation in the brain. In contrast, neurons and astrocytes are known to be involved in prion replication and spread. Moreover, certain astrocytes, such as A1 reactive astrocytes, have proven neurotoxic in other neurodegenerative diseases, and thus might also influence the progression of prion-associated neurodegeneration.
Keywords: PLX5622; astroglia; chemokine; cytokine; microglia; neuroinflammation; neuron; prion; scrapie.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Tribouillard-Tanvier D., Race B., Striebel J.F., Carroll J.A., Phillips K., Chesebro B. Early cytokine elevation, PrPres deposition, and gliosis in mouse scrapie: No effect on disease by deletion of cytokine genes IL-12p40 and IL-12p35. J. Virol. 2012;86:10377–10383. doi: 10.1128/JVI.01340-12. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
