

NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors
- PMID: 30651290
- PMCID: PMC7906796
- DOI: 10.1158/2326-6066.CIR-18-0572
NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors
Abstract
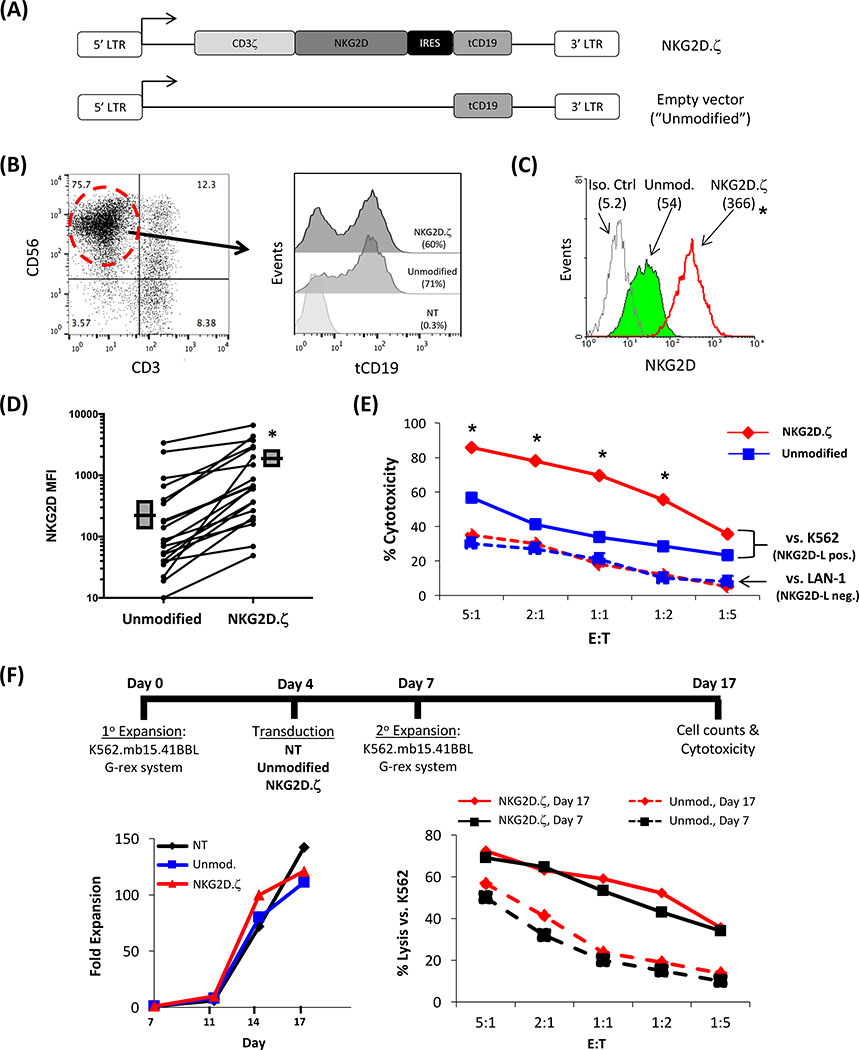
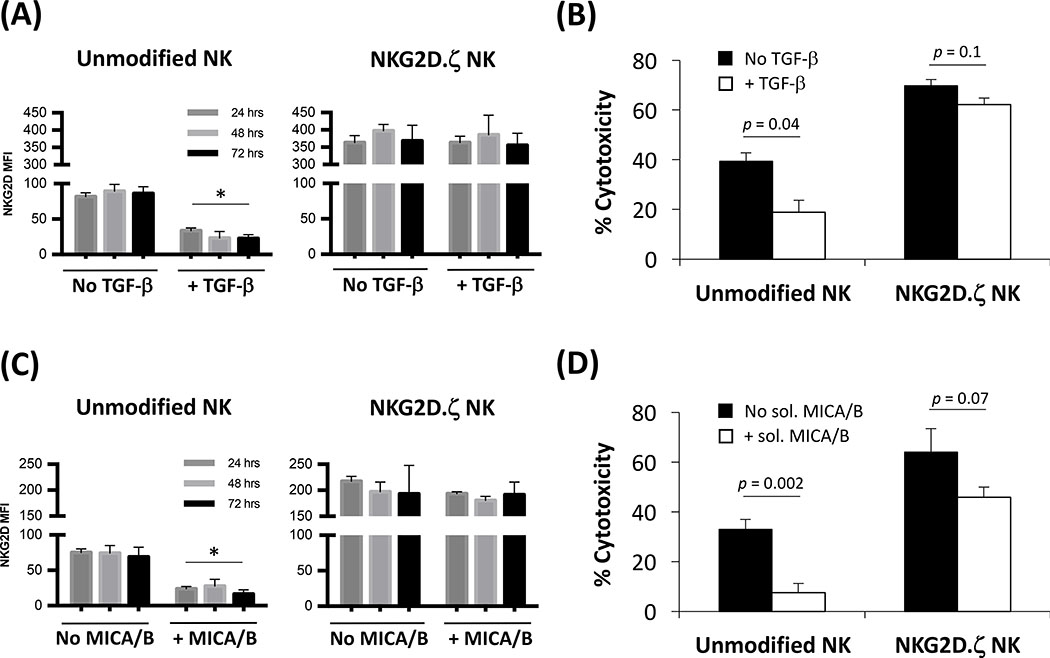
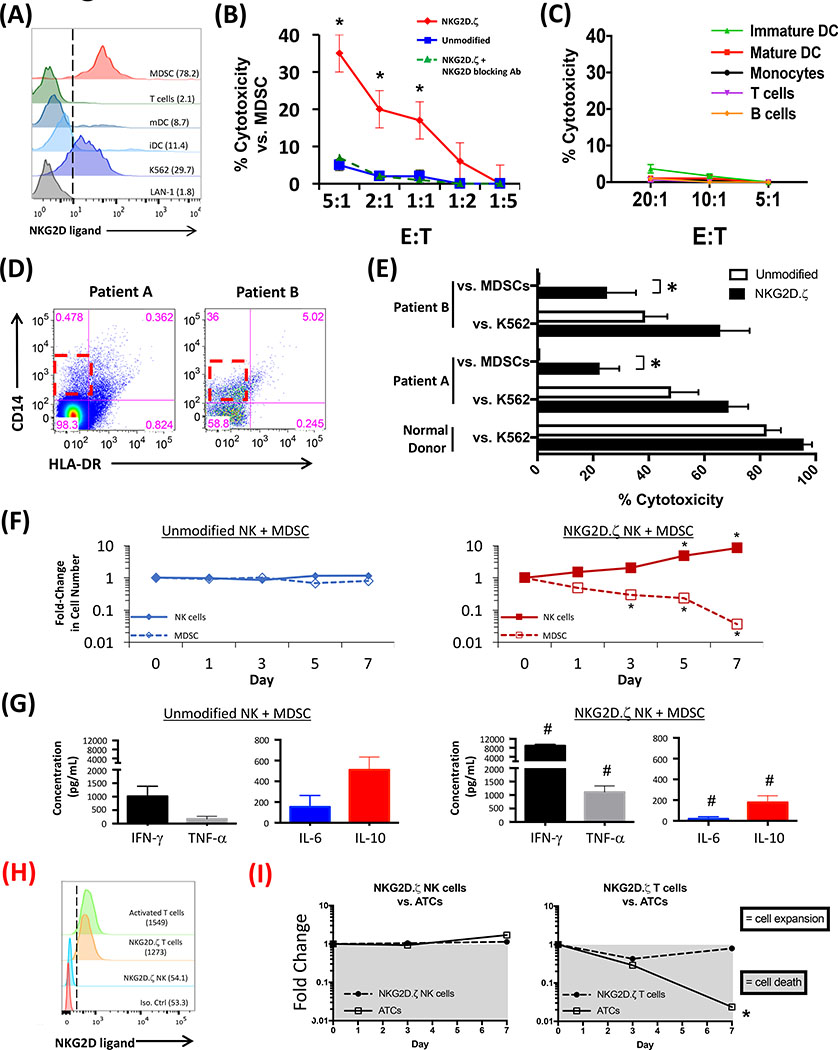
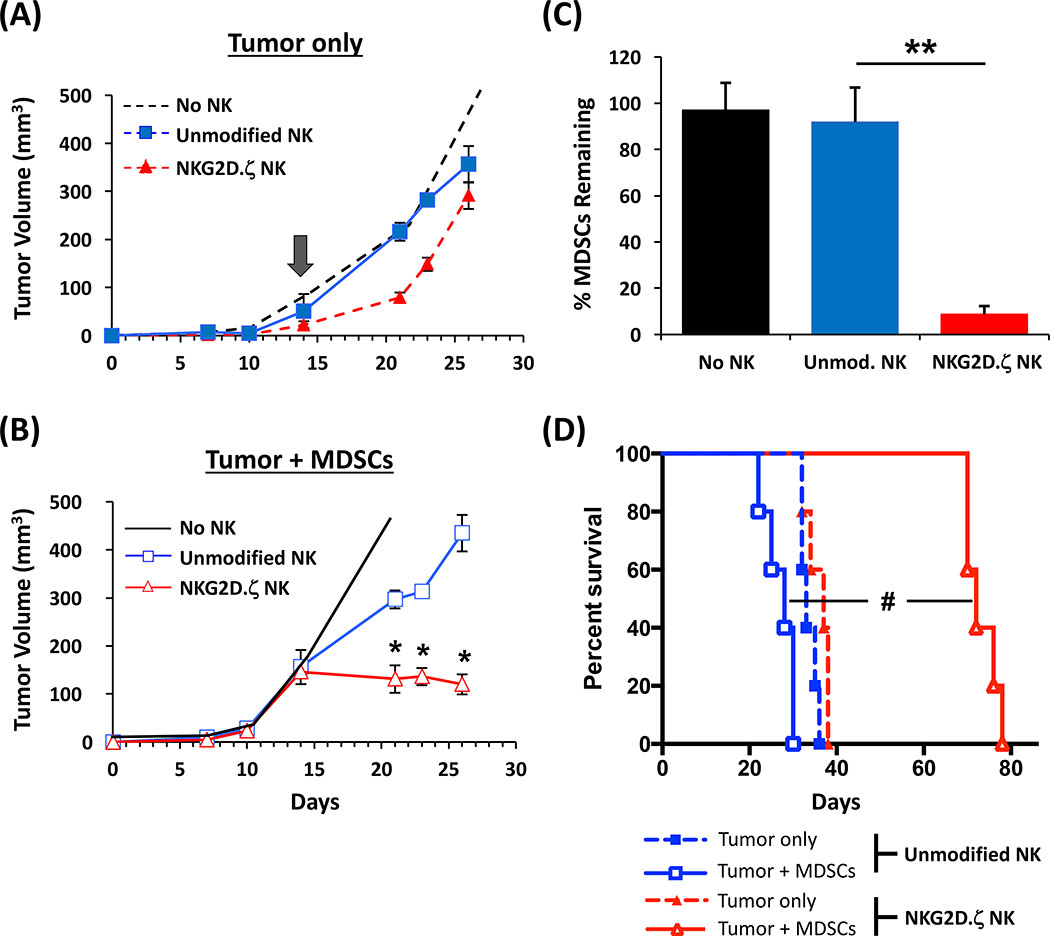
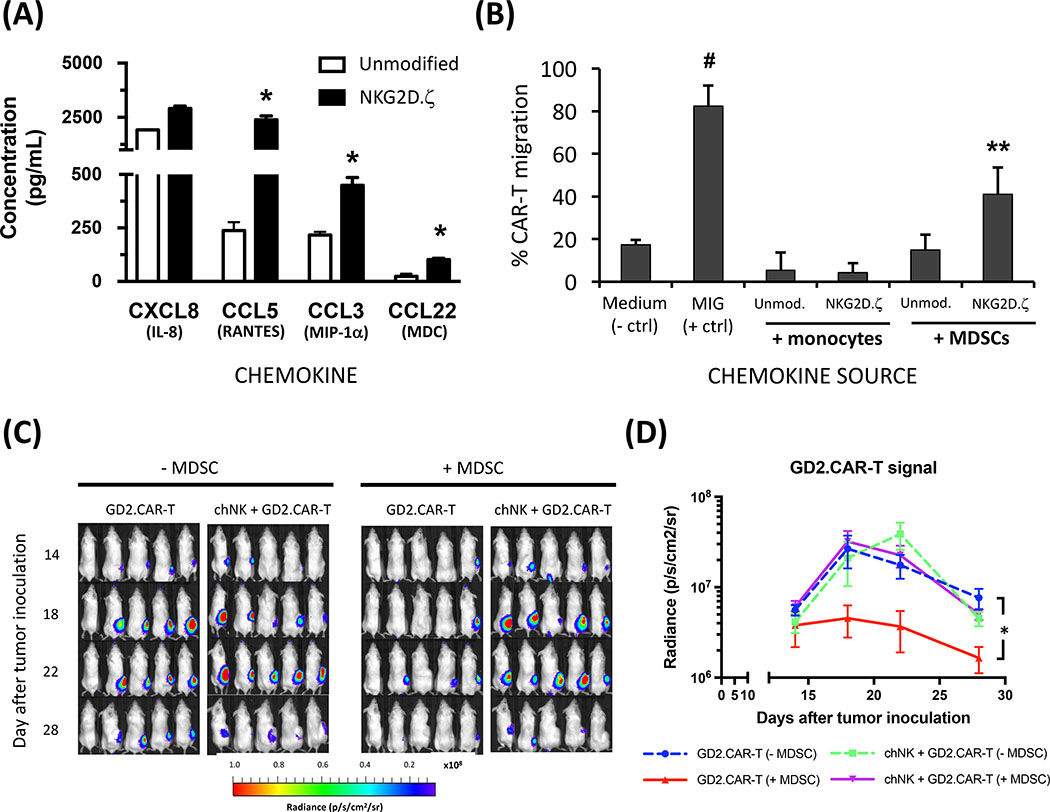
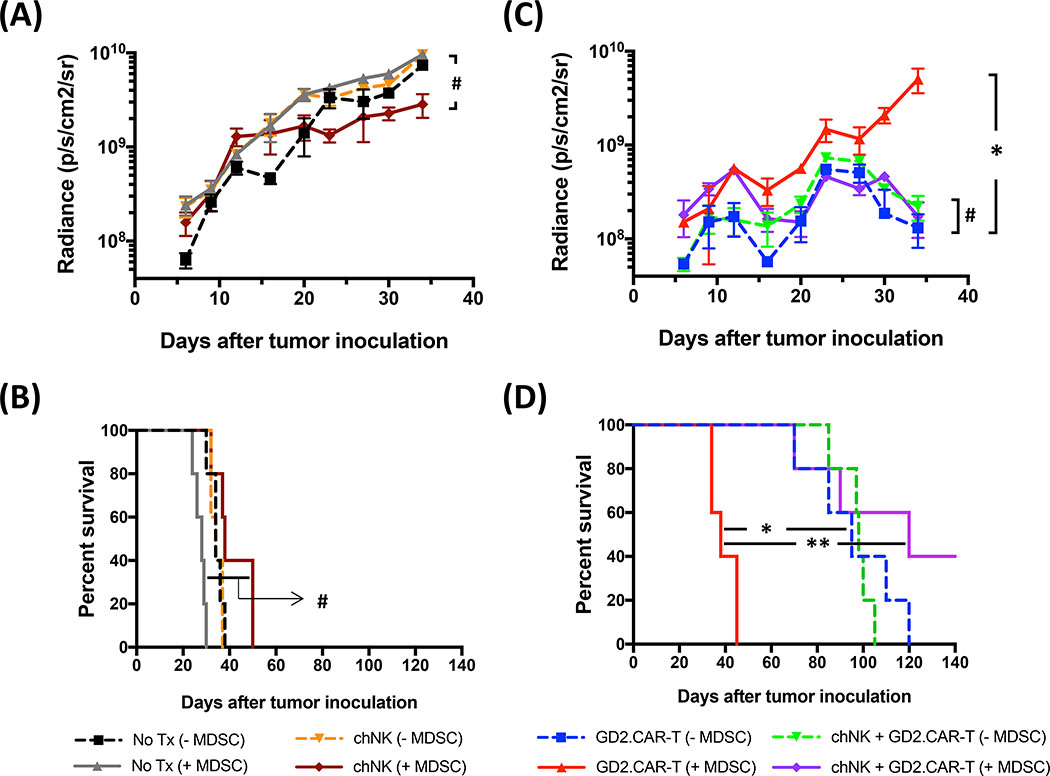
Solid tumors are refractory to cellular immunotherapies in part because they contain suppressive immune effectors such as myeloid-derived suppressor cells (MDSCs) that inhibit cytotoxic lymphocytes. Strategies to reverse the suppressive tumor microenvironment (TME) should also attract and activate immune effectors with antitumor activity. To address this need, we developed gene-modified natural killer (NK) cells bearing a chimeric receptor in which the activating receptor NKG2D is fused to the cytotoxic ζ-chain of the T-cell receptor (NKG2D.ζ). NKG2D.ζ-NK cells target MDSCs, which overexpress NKG2D ligands within the TME. We examined the ability of NKG2D.ζ-NK cells to eliminate MDSCs in a xenograft TME model and improve the antitumor function of tumor-directed chimeric antigen receptor (CAR)-modified T cells. We show that NKG2D.ζ-NK cells are cytotoxic against MDSCs, but spare NKG2D ligand-expressing normal tissues. NKG2D.ζ-NK cells, but not unmodified NK cells, secrete proinflammatory cytokines and chemokines in response to MDSCs at the tumor site and improve infiltration and antitumor activity of subsequently infused CAR-T cells, even in tumors for which an immunosuppressive TME is an impediment to treatment. Unlike endogenous NKG2D, NKG2D.ζ is not susceptible to TME-mediated downmodulation and thus maintains its function even within suppressive microenvironments. As clinical confirmation, NKG2D.ζ-NK cells generated from patients with neuroblastoma killed autologous intratumoral MDSCs capable of suppressing CAR-T function. A combination therapy for solid tumors that includes both NKG2D.ζ-NK cells and CAR-T cells may improve responses over therapies based on CAR-T cells alone.
©2019 American Association for Cancer Research.
Conflict of interest statement
The authors declare no potential conflicts of interest.
Figures
References
-
- Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, Byatte AJ, Kirillova N, Valle JW, Sharma SK, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer immunology, immunotherapy : CII. 2017;66(11):1425–36. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
