

The Nonstructural NS1 Protein of Influenza Viruses Modulates TP53 Splicing through Host Factor CPSF4
- PMID: 30651364
- PMCID: PMC6430556
- DOI: 10.1128/JVI.02168-18
The Nonstructural NS1 Protein of Influenza Viruses Modulates TP53 Splicing through Host Factor CPSF4
Abstract
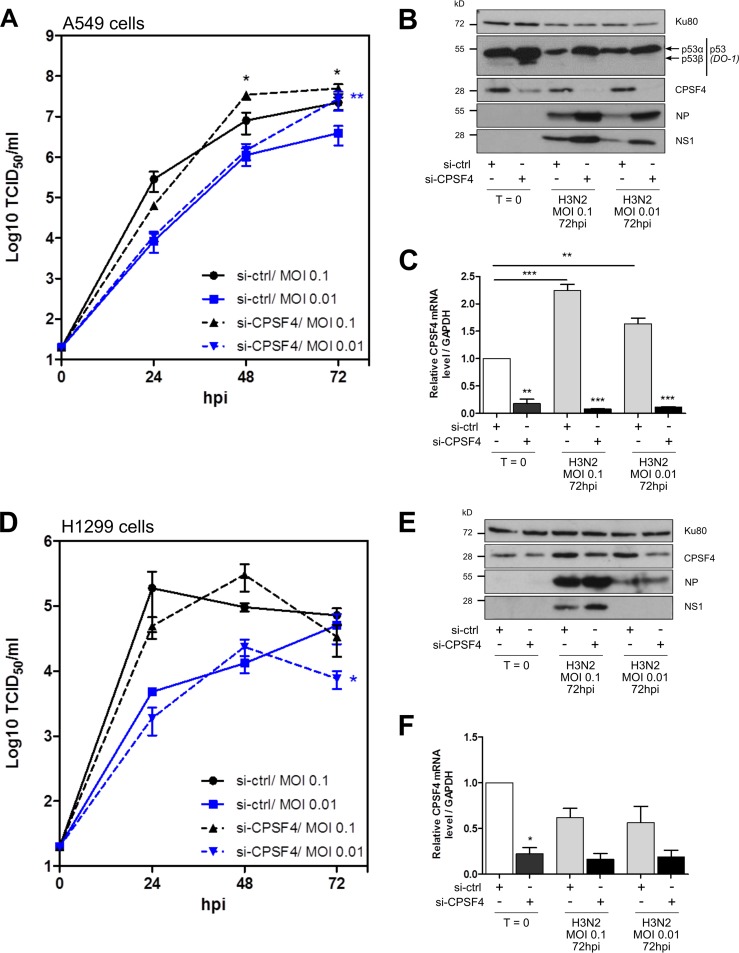
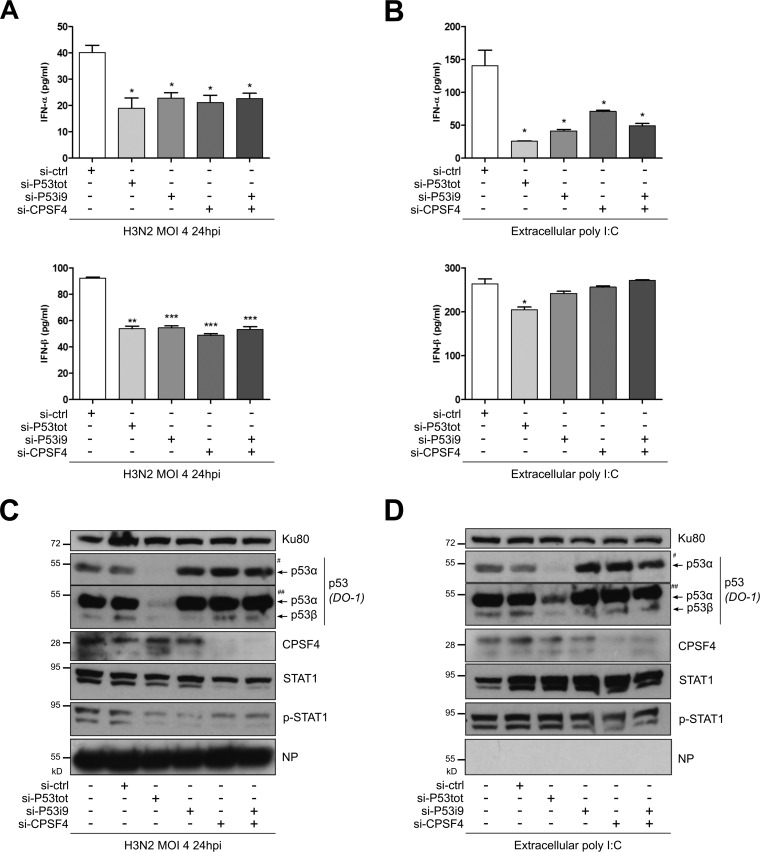
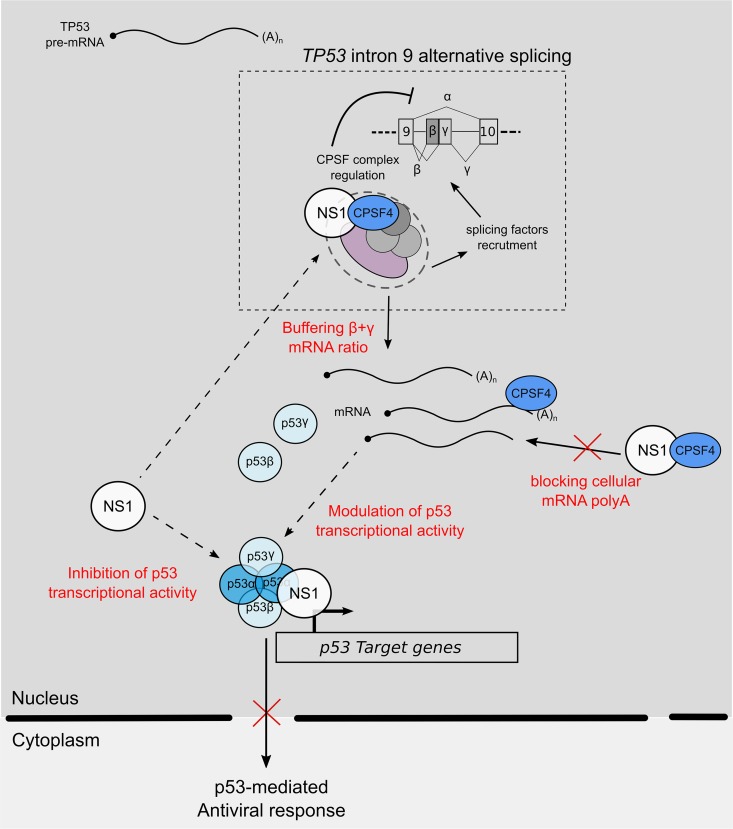
Influenza A viruses (IAV) are known to modulate and "hijack" several cellular host mechanisms, including gene splicing and RNA maturation machineries. These modulations alter host cellular responses and enable an optimal expression of viral products throughout infection. The interplay between the host protein p53 and IAV, in particular through the viral nonstructural protein NS1, has been shown to be supportive for IAV replication. However, it remains unknown whether alternatively spliced isoforms of p53, known to modulate p53 transcriptional activity, are affected by IAV infection and contribute to IAV replication. Using a TP53 minigene, which mimics intron 9 alternative splicing, we have shown here that the NS1 protein of IAV changes the expression pattern of p53 isoforms. Our results demonstrate that CPSF4 (cellular protein cleavage and polyadenylation specificity factor 4) independently and the interaction between NS1 and CPSF4 modulate the alternative splicing of TP53 transcripts, which may result in the differential activation of p53-responsive genes. Finally, we report that CPSF4 and most likely beta and gamma spliced p53 isoforms affect both viral replication and IAV-associated type I interferon secretion. All together, our data show that cellular p53 and CPSF4 factors, both interacting with viral NS1, have a crucial role during IAV replication that allows IAV to interact with and alter the expression of alternatively spliced p53 isoforms in order to regulate the cellular innate response, especially via type I interferon secretion, and perform efficient viral replication.IMPORTANCE Influenza A viruses (IAV) constitute a major public health issue, causing illness and death in high-risk populations during seasonal epidemics or pandemics. IAV are known to modulate cellular pathways to promote their replication and avoid immune restriction via the targeting of several cellular proteins. One of these proteins, p53, is a master regulator involved in a large panel of biological processes, including cell cycle arrest, apoptosis, or senescence. This "cellular gatekeeper" is also involved in the control of viral infections, and viruses have developed a wide diversity of mechanisms to modulate/hijack p53 functions to achieve an optimal replication in their hosts. Our group and others have previously shown that p53 activity is finely modulated by different multilevel mechanisms during IAV infection. Here, we characterized IAV nonstructural protein NS1 and the cellular factor CPSF4 as major partners involved in the IAV-induced modulation of the TP53 alternative splicing that was associated with a strong modulation of p53 activity and notably the p53-mediated antiviral response.
Keywords: CPSF30; antiviral response; influenza viruses; p53; splicing; virus-host interactions.
Copyright © 2019 American Society for Microbiology.
Figures
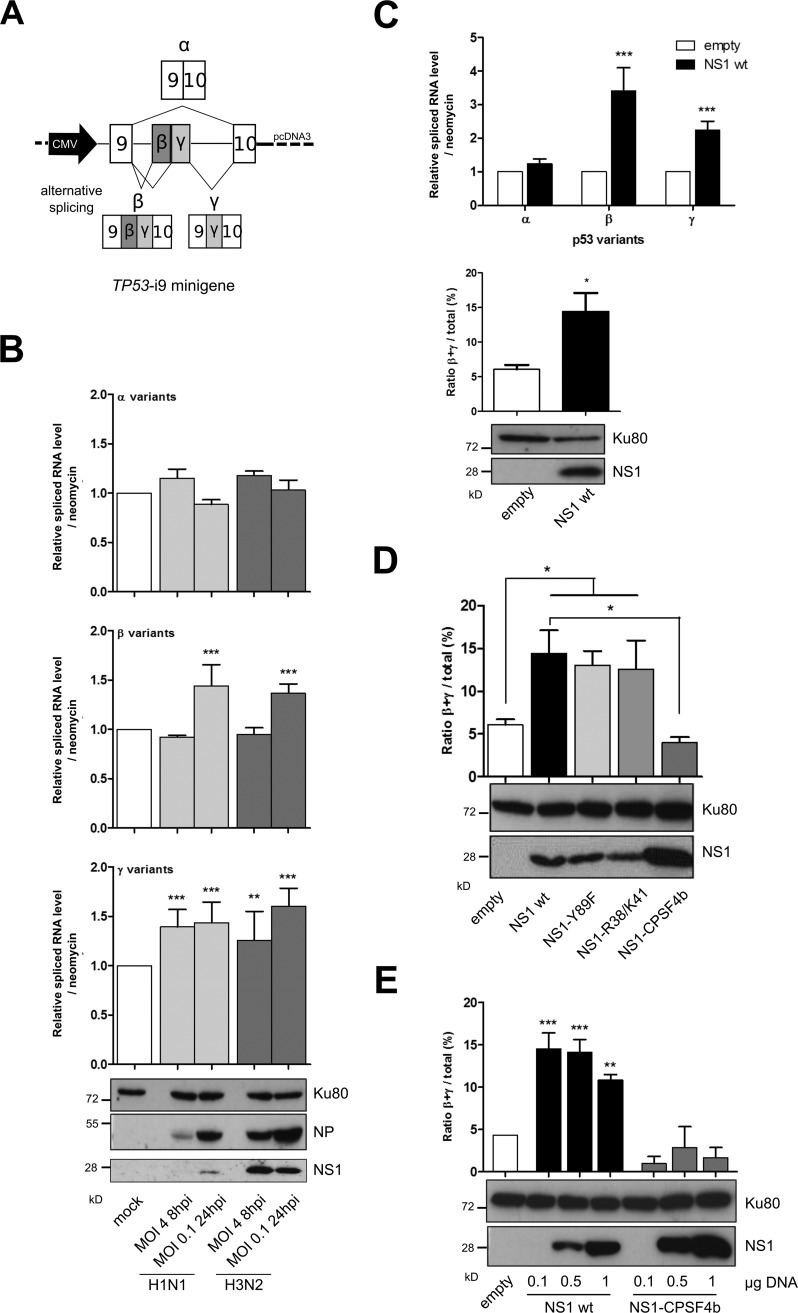
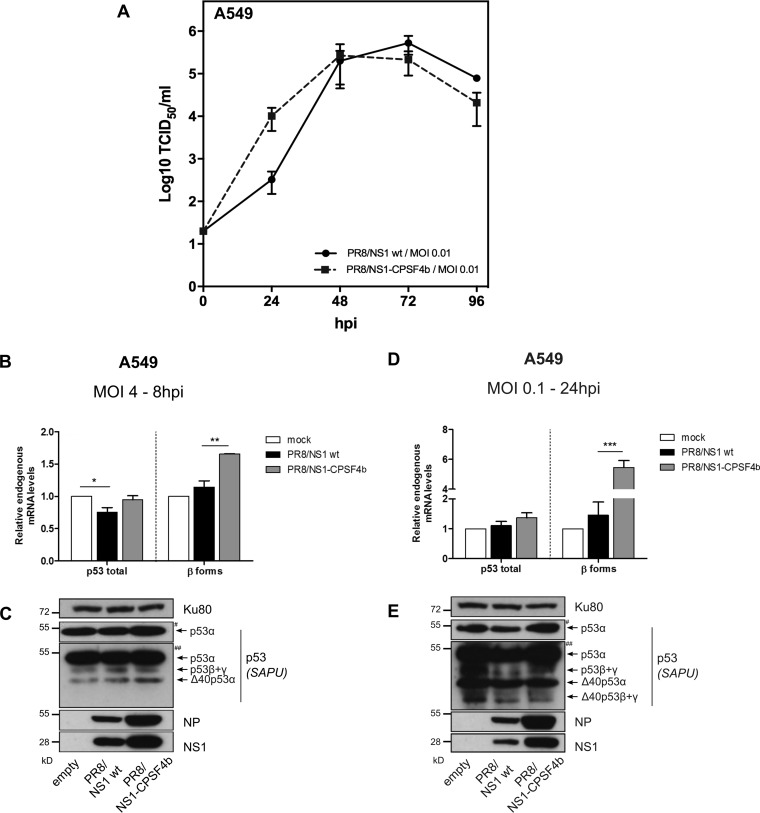
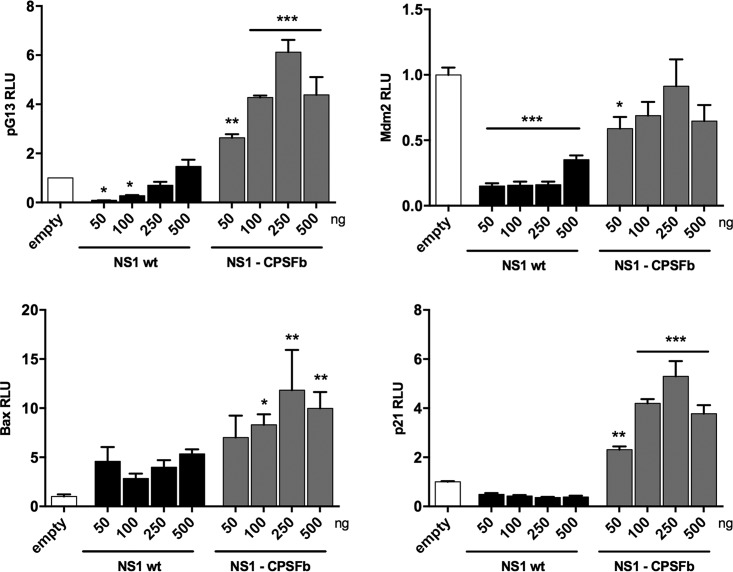
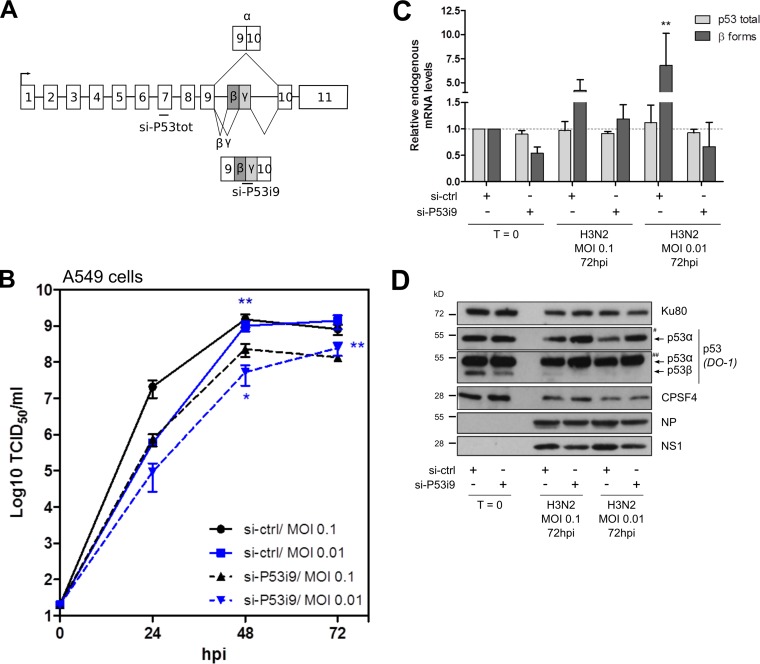
Similar articles
-
Modulation of Innate Immune Responses by the Influenza A NS1 and PA-X Proteins.Viruses. 2018 Dec 12;10(12):708. doi: 10.3390/v10120708. Viruses. 2018. PMID: 30545063 Free PMC article. Review.
-
Differential Modulation of Innate Immune Responses in Human Primary Cells by Influenza A Viruses Carrying Human or Avian Nonstructural Protein 1.J Virol. 2019 Dec 12;94(1):e00999-19. doi: 10.1128/JVI.00999-19. Print 2019 Dec 12. J Virol. 2019. PMID: 31597767 Free PMC article.
-
Mammalian Adaptation of an Avian Influenza A Virus Involves Stepwise Changes in NS1.J Virol. 2018 Feb 12;92(5):e01875-17. doi: 10.1128/JVI.01875-17. Print 2018 Mar 1. J Virol. 2018. PMID: 29237841 Free PMC article.
-
Interplay of PA-X and NS1 Proteins in Replication and Pathogenesis of a Temperature-Sensitive 2009 Pandemic H1N1 Influenza A Virus.J Virol. 2017 Aug 10;91(17):e00720-17. doi: 10.1128/JVI.00720-17. Print 2017 Sep 1. J Virol. 2017. PMID: 28637750 Free PMC article.
-
Immunomodulatory Nonstructural Proteins of Influenza A Viruses.Trends Microbiol. 2018 Jul;26(7):624-636. doi: 10.1016/j.tim.2017.12.006. Epub 2018 Jan 17. Trends Microbiol. 2018. PMID: 29373257 Review.
Cited by
-
The applications of live attenuated influenza a virus with modified NS1 gene.Mol Ther Nucleic Acids. 2025 Feb 1;36(1):102471. doi: 10.1016/j.omtn.2025.102471. eCollection 2025 Mar 11. Mol Ther Nucleic Acids. 2025. PMID: 40046952 Free PMC article. Review.
-
Abnormal global alternative RNA splicing in COVID-19 patients.PLoS Genet. 2022 Apr 14;18(4):e1010137. doi: 10.1371/journal.pgen.1010137. eCollection 2022 Apr. PLoS Genet. 2022. PMID: 35421082 Free PMC article.
-
RNA virus infections and their effect on host alternative splicing.Antiviral Res. 2023 Feb;210:105503. doi: 10.1016/j.antiviral.2022.105503. Epub 2022 Dec 23. Antiviral Res. 2023. PMID: 36572191 Free PMC article. Review.
-
How Many Mammalian Reovirus Proteins are involved in the Control of the Interferon Response?Pathogens. 2019 Jun 21;8(2):83. doi: 10.3390/pathogens8020083. Pathogens. 2019. PMID: 31234302 Free PMC article. Review.
-
The Central Role of Non-Structural Protein 1 (NS1) in Influenza Biology and Infection.Int J Mol Sci. 2020 Feb 22;21(4):1511. doi: 10.3390/ijms21041511. Int J Mol Sci. 2020. PMID: 32098424 Free PMC article. Review.
References
-
- Paules C, Subbarao K. 2017. Influenza. Lancet 390:697–708. - PubMed
-
- Palese PSM. 2013. Orthomyxoviridae, p 1151–1185. In Knipe DM, Howley P (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
