

Antigenicity and Immunogenicity of Differentially Glycosylated Hepatitis C Virus E2 Envelope Proteins Expressed in Mammalian and Insect Cells
- PMID: 30651366
- PMCID: PMC6430559
- DOI: 10.1128/JVI.01403-18
Antigenicity and Immunogenicity of Differentially Glycosylated Hepatitis C Virus E2 Envelope Proteins Expressed in Mammalian and Insect Cells
Abstract
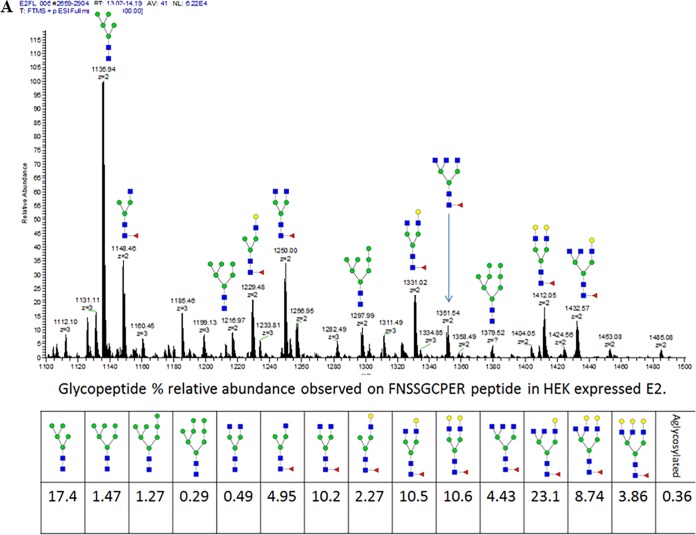
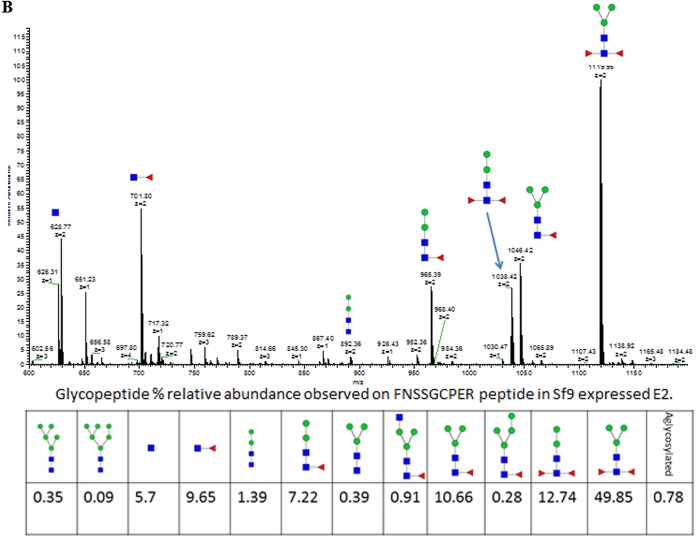
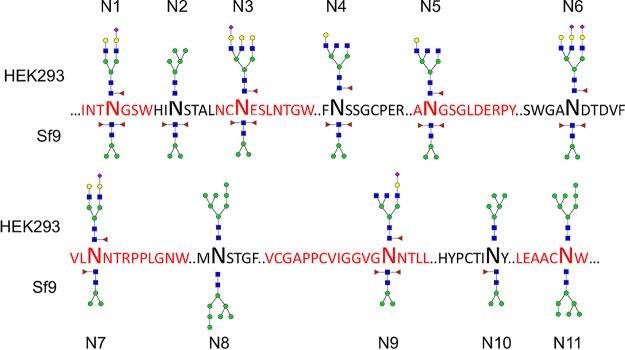
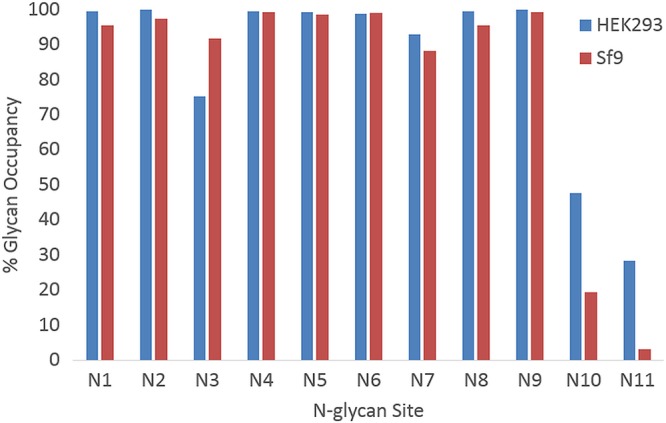
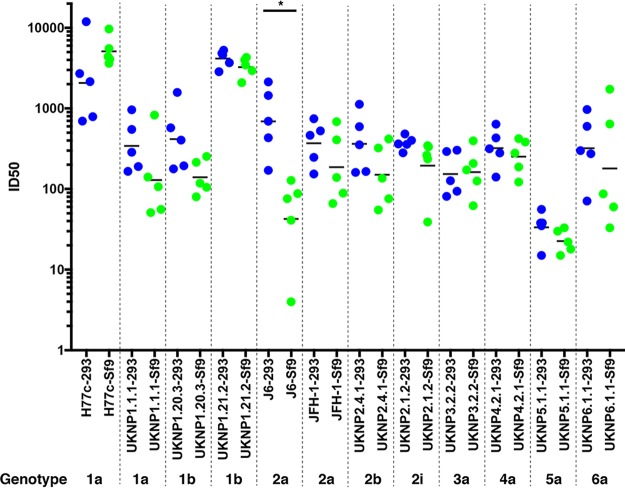
The development of a prophylactic vaccine for hepatitis C virus (HCV) remains a global health challenge. Cumulative evidence supports the importance of antibodies targeting the HCV E2 envelope glycoprotein to facilitate viral clearance. However, a significant challenge for a B cell-based vaccine is focusing the immune response on conserved E2 epitopes capable of eliciting neutralizing antibodies not associated with viral escape. We hypothesized that glycosylation might influence the antigenicity and immunogenicity of E2. Accordingly, we performed head-to-head molecular, antigenic, and immunogenic comparisons of soluble E2 (sE2) produced in (i) mammalian (HEK293) cells, which confer mostly complex- and high-mannose-type glycans; and (ii) insect (Sf9) cells, which impart mainly paucimannose-type glycans. Mass spectrometry demonstrated that all 11 predicted N-glycosylation sites were utilized in both HEK293- and Sf9-derived sE2, but that N-glycans in insect sE2 were on average smaller and less complex. Both proteins bound CD81 and were recognized by conformation-dependent antibodies. Mouse immunogenicity studies revealed that similar polyclonal antibody responses were generated against antigenic domains A to E of E2. Although neutralizing antibody titers showed that Sf9-derived sE2 induced moderately stronger responses than did HEK293-derived sE2 against the homologous HCV H77c isolate, the two proteins elicited comparable neutralization titers against heterologous isolates. Given that global alteration of HCV E2 glycosylation by expression in different hosts did not appreciably affect antigenicity or overall immunogenicity, a more productive approach to increasing the antibody response to neutralizing epitopes may be complete deletion, rather than just modification, of specific N-glycans proximal to these epitopes.IMPORTANCE The development of a vaccine for hepatitis C virus (HCV) remains a global health challenge. A major challenge for vaccine development is focusing the immune response on conserved regions of the HCV envelope protein, E2, capable of eliciting neutralizing antibodies. Modification of E2 by glycosylation might influence the immunogenicity of E2. Accordingly, we performed molecular and immunogenic comparisons of E2 produced in mammalian and insect cells. Mass spectrometry demonstrated that the predicted glycosylation sites were utilized in both mammalian and insect cell E2, although the glycan types in insect cell E2 were smaller and less complex. Mouse immunogenicity studies revealed similar polyclonal antibody responses. However, insect cell E2 induced stronger neutralizing antibody responses against the homologous isolate used in the vaccine, albeit the two proteins elicited comparable neutralization titers against heterologous isolates. A more productive approach for vaccine development may be complete deletion of specific glycans in the E2 protein.
Keywords: HCV; envelope glycoproteins; hepatitis C virus; immunogenicity; insect cells; mammalian cells; neutralization; vaccine.
Copyright © 2019 Urbanowicz et al.
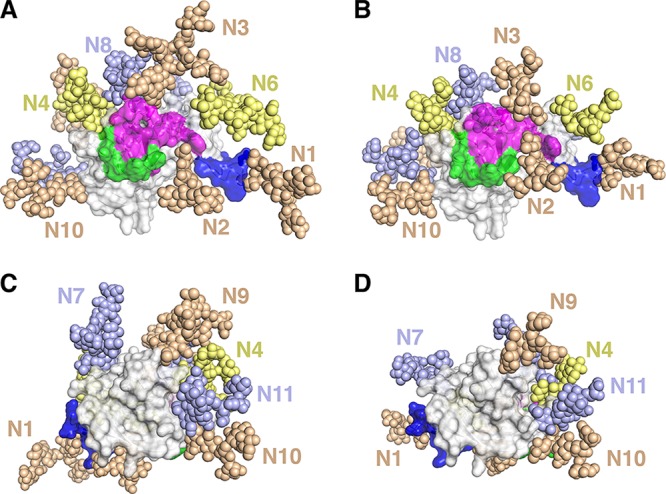
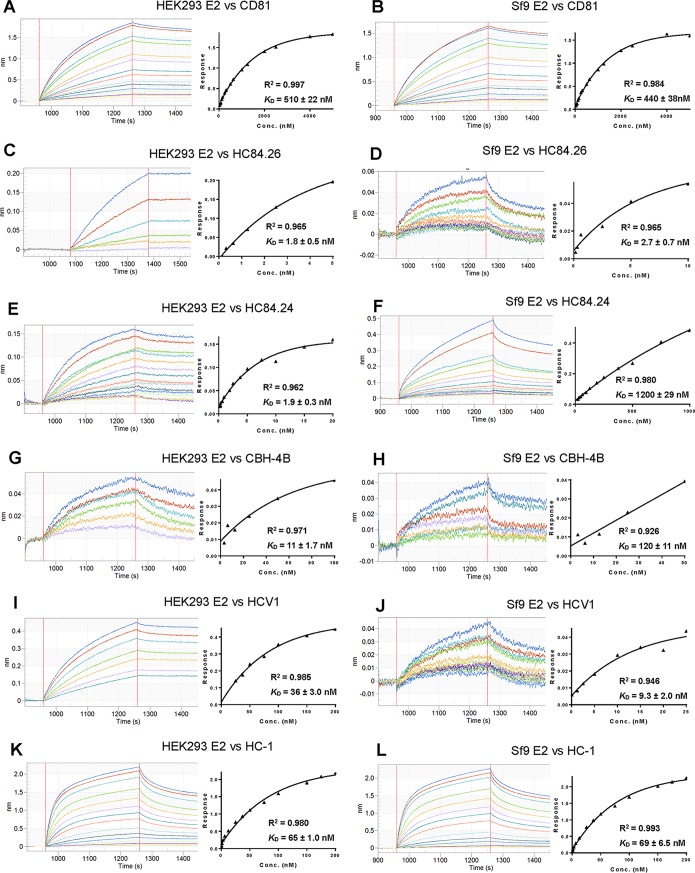
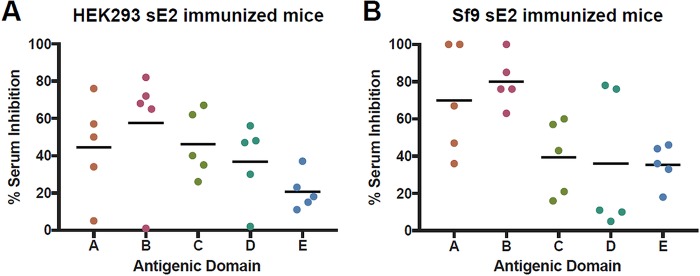
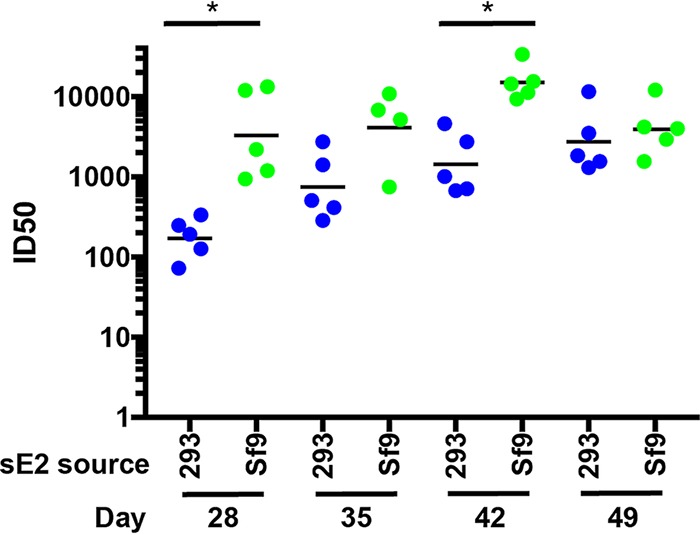
Figures

Similar articles
-
Structure-Based Design of Hepatitis C Virus E2 Glycoprotein Improves Serum Binding and Cross-Neutralization.J Virol. 2020 Oct 27;94(22):e00704-20. doi: 10.1128/JVI.00704-20. Print 2020 Oct 27. J Virol. 2020. PMID: 32878891 Free PMC article.
-
Altered Glycosylation Patterns Increase Immunogenicity of a Subunit Hepatitis C Virus Vaccine, Inducing Neutralizing Antibodies Which Confer Protection in Mice.J Virol. 2016 Nov 14;90(23):10486-10498. doi: 10.1128/JVI.01462-16. Print 2016 Dec 1. J Virol. 2016. PMID: 27630242 Free PMC article.
-
Functional and immunogenic characterization of diverse HCV glycoprotein E2 variants.J Hepatol. 2019 Apr;70(4):593-602. doi: 10.1016/j.jhep.2018.11.003. Epub 2018 Nov 13. J Hepatol. 2019. PMID: 30439392
-
Glycan Shielding and Modulation of Hepatitis C Virus Neutralizing Antibodies.Front Immunol. 2018 Apr 27;9:910. doi: 10.3389/fimmu.2018.00910. eCollection 2018. Front Immunol. 2018. PMID: 29755477 Free PMC article. Review.
-
Neutralizing antibody response to hepatitis C virus.Viruses. 2011 Nov;3(11):2127-45. doi: 10.3390/v3112127. Epub 2011 Nov 2. Viruses. 2011. PMID: 22163337 Free PMC article. Review.
Cited by
-
Production and immunogenicity of different prophylactic vaccines for hepatitis C virus (Review).Exp Ther Med. 2022 May 30;24(1):474. doi: 10.3892/etm.2022.11401. eCollection 2022 Jul. Exp Ther Med. 2022. PMID: 35761816 Free PMC article. Review.
-
Impact of Protein Glycosylation on the Design of Viral Vaccines.Adv Biochem Eng Biotechnol. 2021;175:319-354. doi: 10.1007/10_2020_132. Adv Biochem Eng Biotechnol. 2021. PMID: 32935143 Review.
-
Supramolecular assembly of Toll-like receptor 7/8 agonist into multimeric water-soluble constructs enables superior immune stimulation in vitro and in vivo.ACS Appl Bio Mater. 2020 May 18;3(5):3187-3195. doi: 10.1021/acsabm.0c00189. Epub 2020 Apr 8. ACS Appl Bio Mater. 2020. PMID: 33880435 Free PMC article.
-
Inactivated genotype 1a, 2a and 3a HCV vaccine candidates induced broadly neutralising antibodies in mice.Gut. 2023 Mar;72(3):560-572. doi: 10.1136/gutjnl-2021-326323. Epub 2022 Aug 2. Gut. 2023. PMID: 35918103 Free PMC article.
-
Analysis of the SARS-CoV-2 spike protein glycan shield: implications for immune recognition.bioRxiv [Preprint]. 2020 May 1:2020.04.07.030445. doi: 10.1101/2020.04.07.030445. bioRxiv. 2020. Update in: Sci Rep. 2020 Sep 14;10(1):14991. doi: 10.1038/s41598-020-71748-7. PMID: 32511307 Free PMC article. Updated. Preprint.
References
-
- Pestka JM, Zeisel MB, Bläser E, Schürmann P, Bartosch B, Cosset FL, Patel AH, Meisel H, Baumert J, Viazov S, Rispeter K, Blum HE, Roggendorf M, Baumert TF. 2007. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci U S A 104:6025–6030. doi:10.1073/pnas.0607026104. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
