

Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms
- PMID: 30651561
- PMCID: PMC6335421
- DOI: 10.1038/s41467-018-08263-x
Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms
Abstract
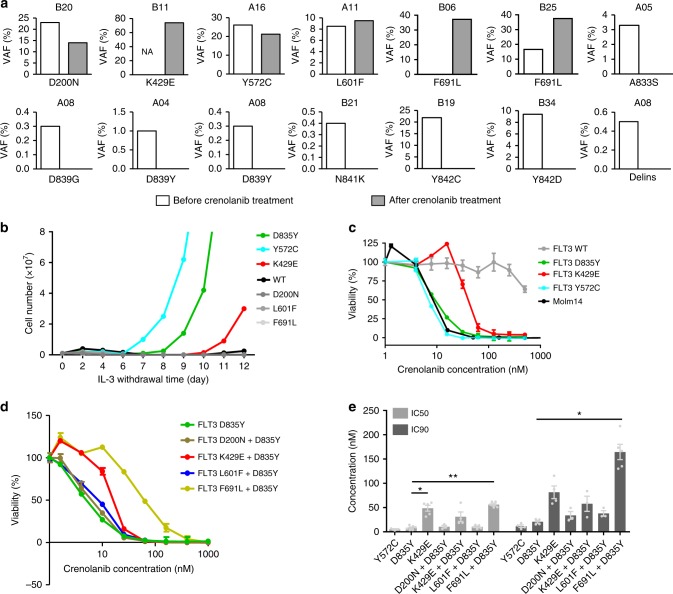
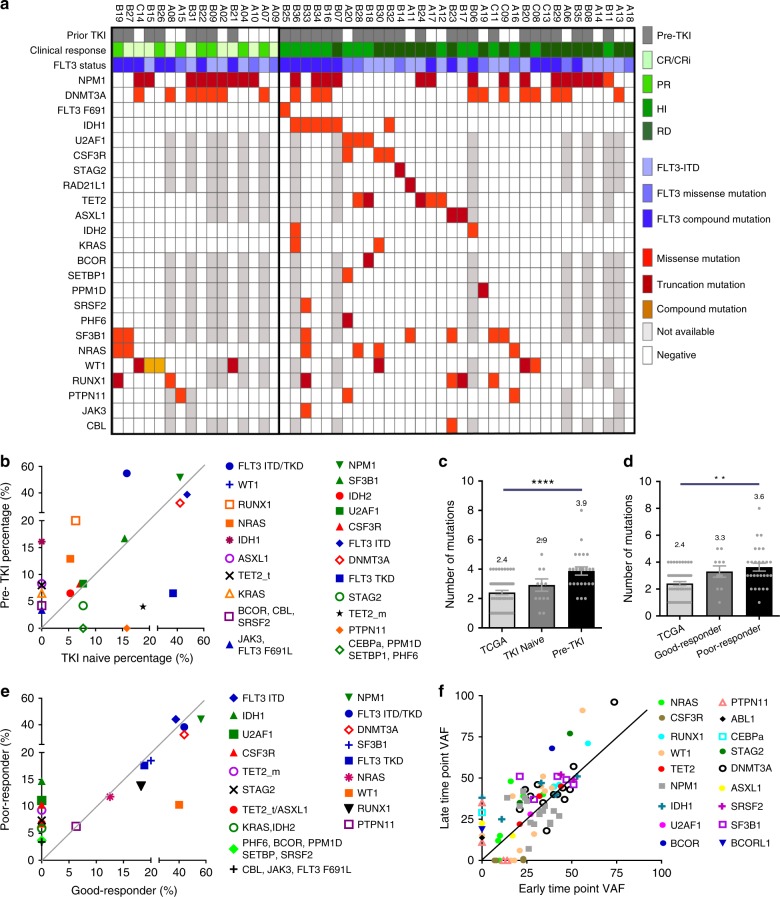
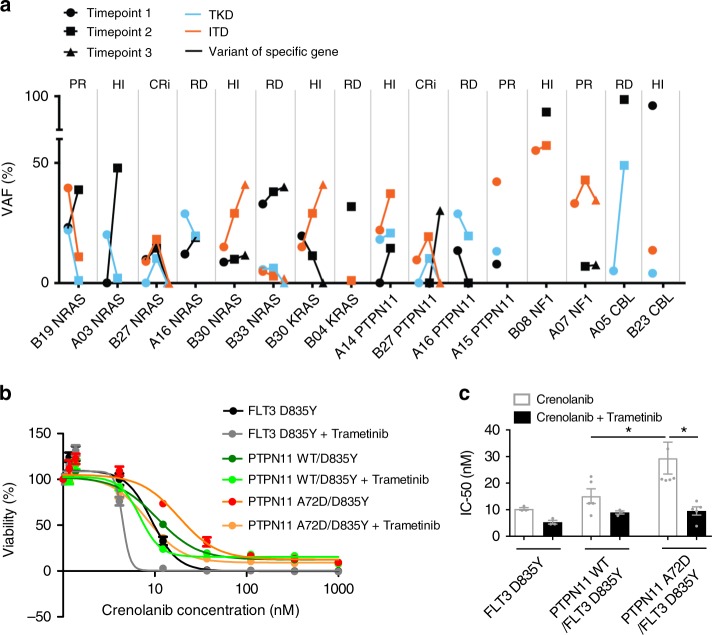
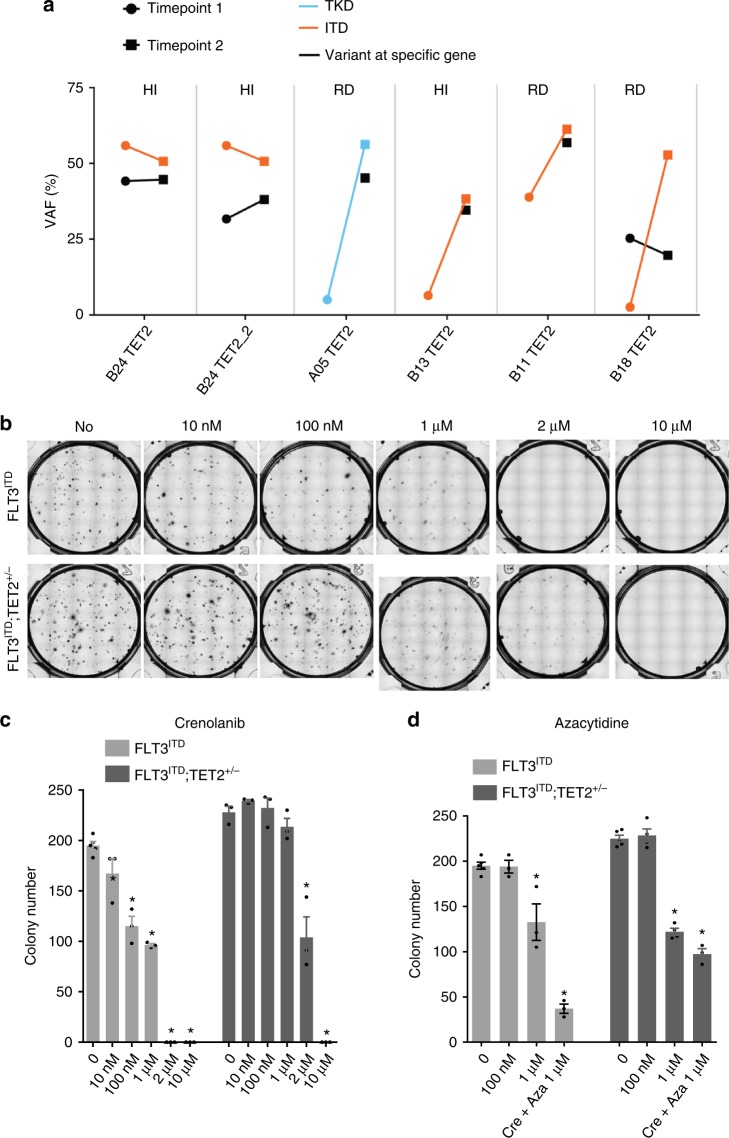
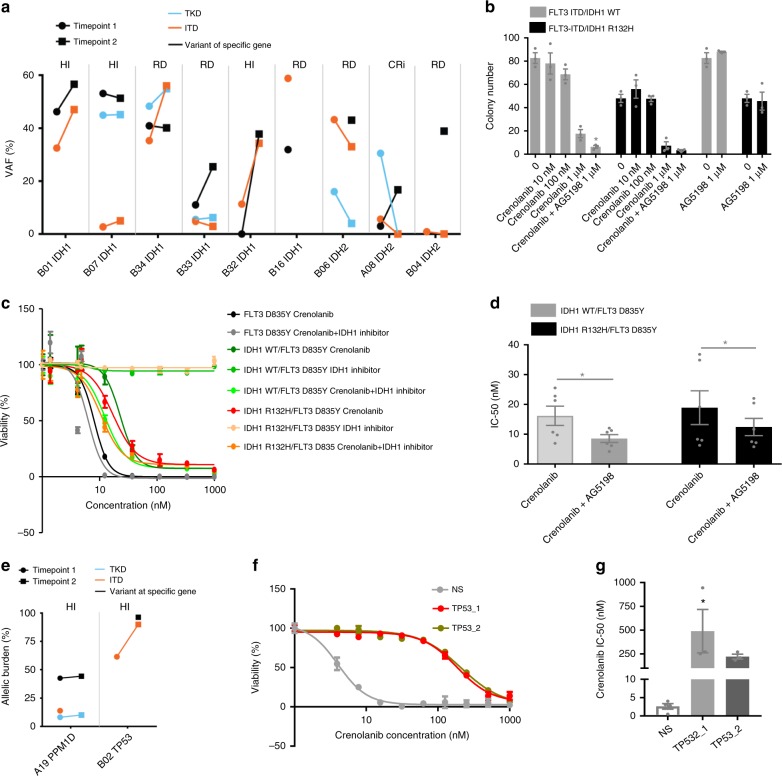
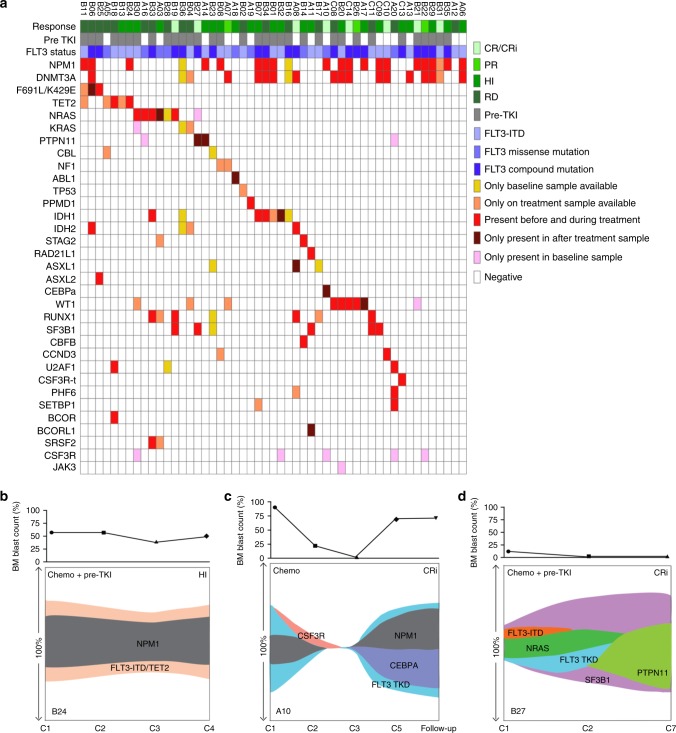
FLT3 mutations are prevalent in AML patients and confer poor prognosis. Crenolanib, a potent type I pan-FLT3 inhibitor, is effective against both internal tandem duplications and resistance-conferring tyrosine kinase domain mutations. While crenolanib monotherapy has demonstrated clinical benefit in heavily pretreated relapsed/refractory AML patients, responses are transient and relapse eventually occurs. Here, to investigate the mechanisms of crenolanib resistance, we perform whole exome sequencing of AML patient samples before and after crenolanib treatment. Unlike other FLT3 inhibitors, crenolanib does not induce FLT3 secondary mutations, and mutations of the FLT3 gatekeeper residue are infrequent. Instead, mutations of NRAS and IDH2 arise, mostly as FLT3-independent subclones, while TET2 and IDH1 predominantly co-occur with FLT3-mutant clones and are enriched in crenolanib poor-responders. The remaining patients exhibit post-crenolanib expansion of mutations associated with epigenetic regulators, transcription factors, and cohesion factors, suggesting diverse genetic/epigenetic mechanisms of crenolanib resistance. Drug combinations in experimental models restore crenolanib sensitivity.
Conflict of interest statement
Research support for J.W.T. is received from Aptose, Array, AstraZeneca, Constellation, Genentech, Gilead, Incyte, Janssen, Seattle Genetics, Syros, Takeda; co-founder of Vivid Biosciences. B.J.D. potential competing interests-- Scientific Advisory Board: Aileron Therapeutics, ALLCRON, Cepheid, Gilead Sciences, Vivid Biosciences, Celgene & Baxalta (inactive); SAB & Stock: Aptose Biosciences, Blueprint Medicines, Beta Cat, GRAIL, Third Coast Therapeutics, CTI BioPharma (inactive); Scientific Founder & Stock: MolecularMD; Board of Directors & Stock: Amgen; Board of Directors: Burroughs Wellcome Fund, CureOne; Joint Steering Committee: Beat AML LLS; Clinical Trial Funding: Novartis, Bristol-Myers Squibb, Pfizer; Royalties from Patent 6958335 (Novartis exclusive license) and OHSU and Dana-Farber Cancer Institute (one Merck exclusive license) H.H., Y.L.L., R.S., C.F. and V.J. are employees of AROG Pharmaceuticals. The remaining authors declare no competing interests.
Figures
References
-
- Kottaridis PD, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United King. Blood. 2001;98:1752–1759. doi: 10.1182/blood.V98.6.1752. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
