

Neuroprotective Mitochondrial Remodeling by AKAP121/PKA Protects HT22 Cell from Glutamate-Induced Oxidative Stress
- PMID: 30652267
- PMCID: PMC6615994
- DOI: 10.1007/s12035-018-1464-3
Neuroprotective Mitochondrial Remodeling by AKAP121/PKA Protects HT22 Cell from Glutamate-Induced Oxidative Stress
Abstract
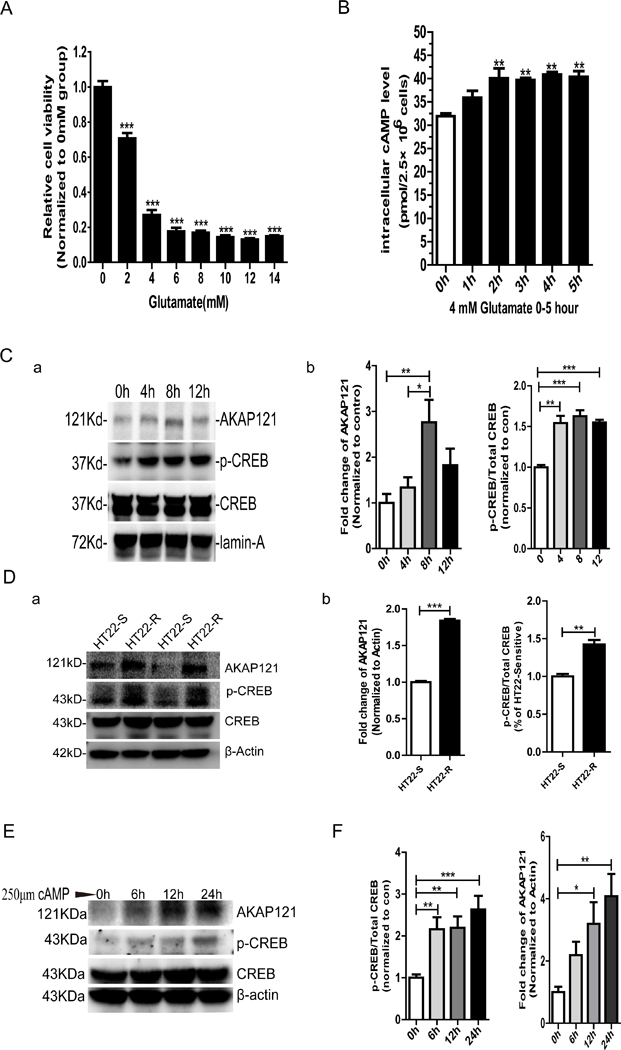
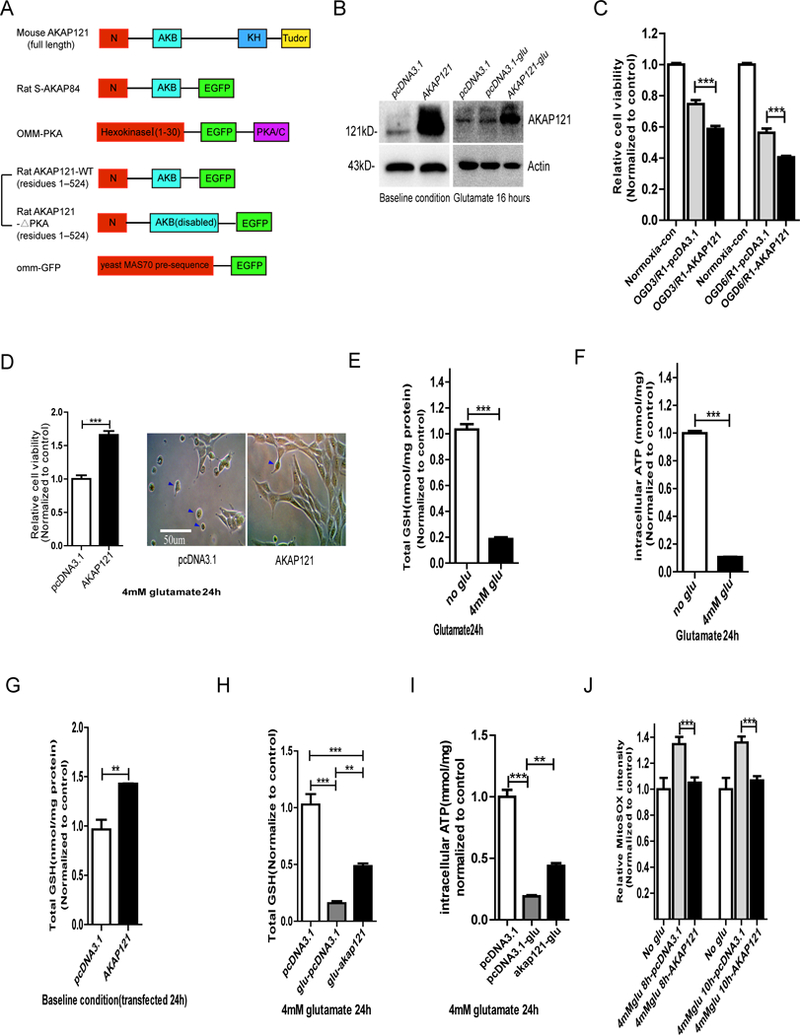
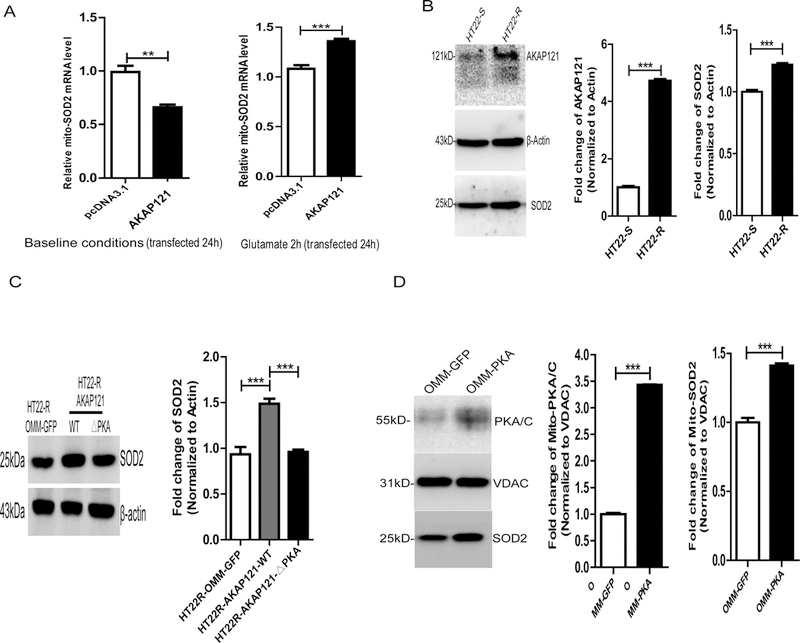
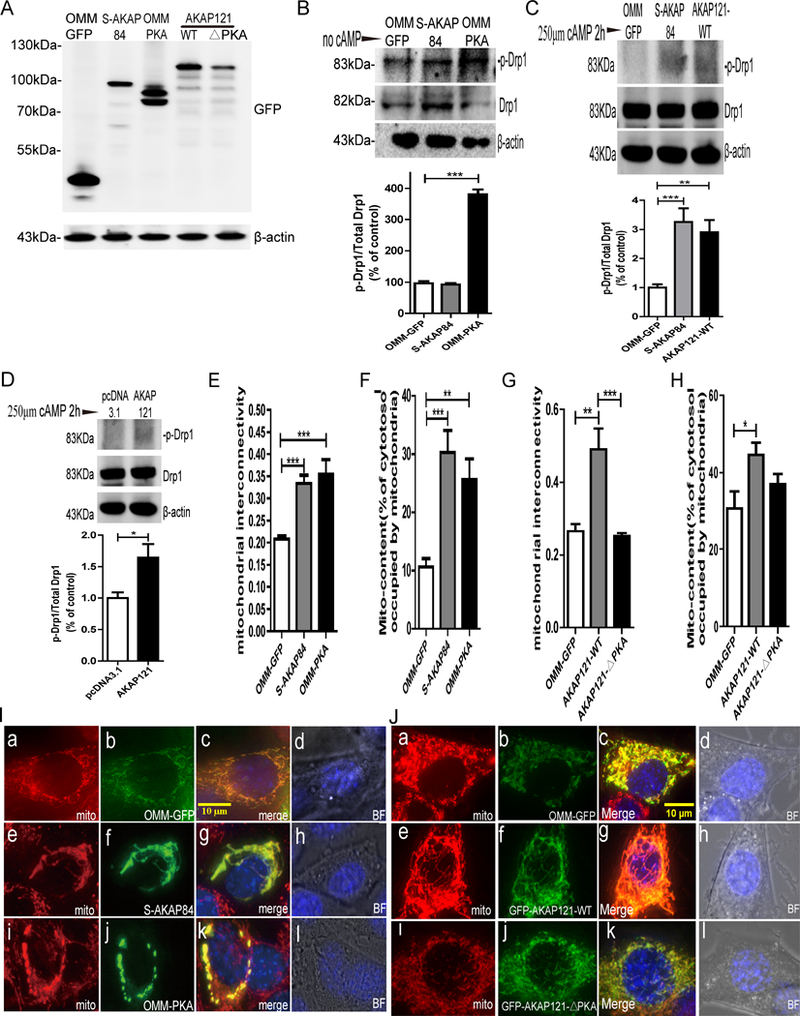
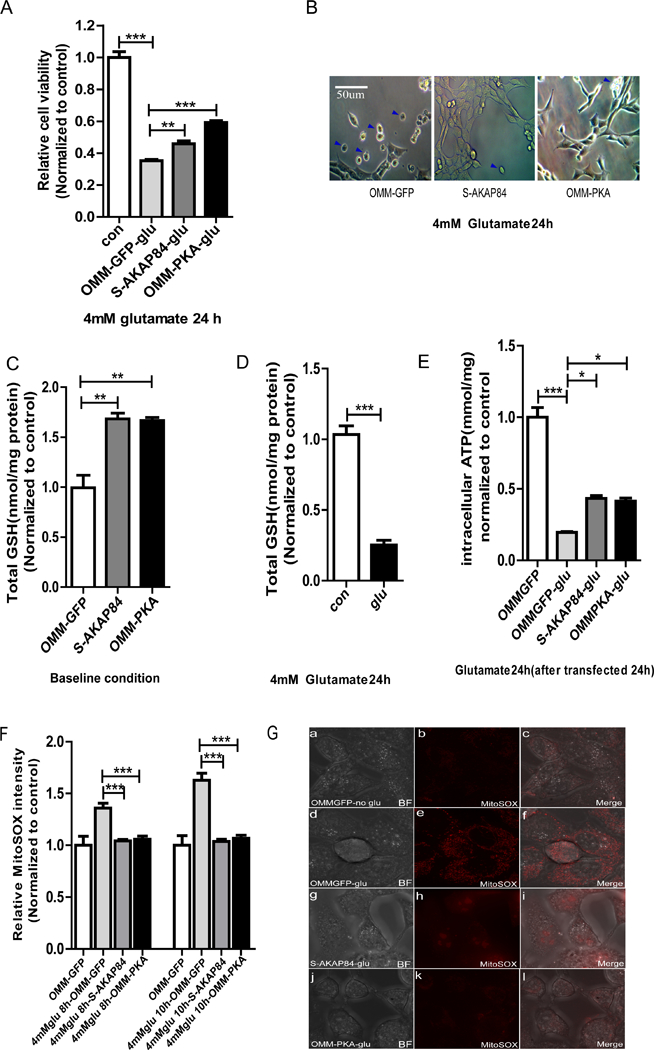
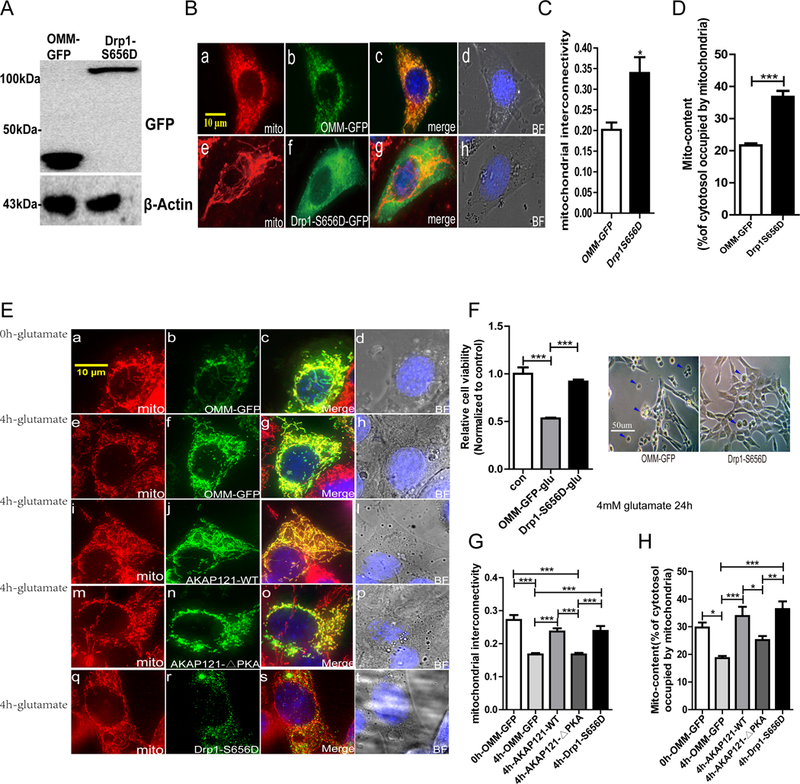
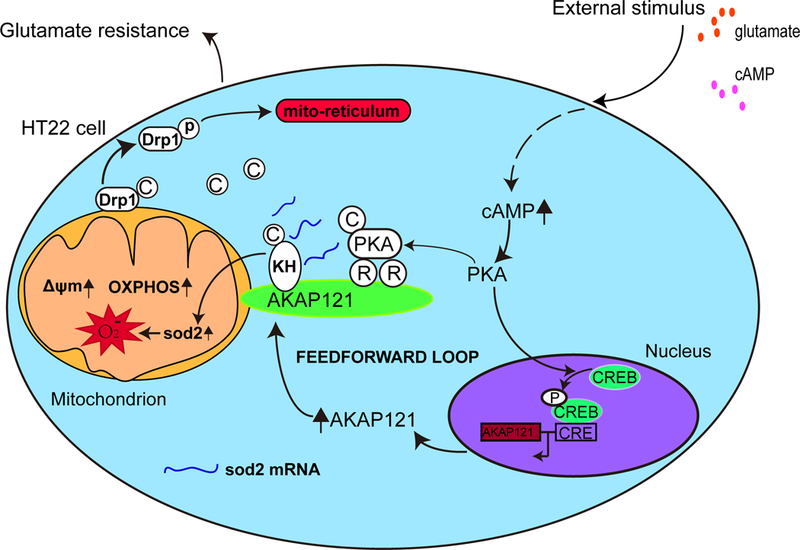
Protein kinase A (PKA) is a ser/thr kinase that is critical for maintaining essential neuronal functions including mitochondrial homeostasis, bioenergetics, neuronal development, and neurotransmission. The endogenous pool of PKA is targeted to the mitochondrion by forming a complex with the mitochondrial scaffold A-kinase anchoring protein 121 (AKAP121). Enhanced PKA signaling via AKAP121 leads to PKA-mediated phosphorylation of the fission modulator Drp1, leading to enhanced mitochondrial networks and thereby blocking apoptosis against different toxic insults. In this study, we show for the first time that AKAP121/PKA confers neuroprotection in an in vitro model of oxidative stress induced by exposure to excess glutamate. Unexpectedly, treating mouse hippocampal progenitor neuronal HT22 cells with an acute dose or chronic exposure of glutamate robustly elevates PKA signaling, a beneficial compensatory response that is phenocopied in HT22 cells conditioned to thrive in the presence of excess glutamate but not in parental HT22 cells. Secondly, redirecting the endogenous pool of PKA by transiently transfecting AKAP121 or transfecting a constitutively active mutant of PKA targeted to the mitochondrion (OMM-PKA) or of an isoform of AKAP121 that lacks the KH and Tudor domains (S-AKAP84) are sufficient to significantly block cell death induced by glutamate toxicity but not in an oxygen deprivation/reperfusion model. Conversely, transient transfection of HT22 neuronal cells with a PKA-binding-deficient mutant of AKAP121 is unable to protect against oxidative stress induced by glutamate toxicity suggesting that the catalytic activity of PKA is required for AKAP121's protective effects. Mechanistically, AKAP121 promotes neuroprotection by enhancing PKA-mediated phosphorylation of Drp1 to increase mitochondrial fusion, elevates ATP levels, and elicits an increase in the levels of antioxidants GSH and superoxide dismutase 2 leading to a reduction in the level of mitochondrial superoxide. Overall, our data supports AKAP121/PKA as a new molecular target that confers neuroprotection against glutamate toxicity by phosphorylating Drp1, to stabilize mitochondrial networks and mitochondrial function and to elicit antioxidant responses.
Keywords: A-kinase anchoring protein 121; Dynamin-related protein 1 (Drp1); HT22; Mitochondrion; Oxidative stress; Protein kinase A.
Figures
Similar articles
-
Mitochondrial PKA Is Neuroprotective in a Cell Culture Model of Alzheimer's Disease.Mol Neurobiol. 2021 Jul;58(7):3071-3083. doi: 10.1007/s12035-021-02333-w. Epub 2021 Feb 23. Mol Neurobiol. 2021. PMID: 33624140 Free PMC article.
-
Isoliquiritigenin attenuates glutamate-induced mitochondrial fission via calcineurin-mediated Drp1 dephosphorylation in HT22 hippocampal neuron cells.Neurotoxicology. 2018 Sep;68:133-141. doi: 10.1016/j.neuro.2018.07.011. Epub 2018 Jul 23. Neurotoxicology. 2018. PMID: 30048666
-
Mechanism of neuroprotective mitochondrial remodeling by PKA/AKAP1.PLoS Biol. 2011 Apr;9(4):e1000612. doi: 10.1371/journal.pbio.1000612. Epub 2011 Apr 19. PLoS Biol. 2011. PMID: 21526220 Free PMC article.
-
Role of protein kinase A in regulating mitochondrial function and neuronal development: implications to neurodegenerative diseases.Rev Neurosci. 2015;26(3):359-70. doi: 10.1515/revneuro-2014-0085. Rev Neurosci. 2015. PMID: 25741943 Free PMC article. Review.
-
Pivotal role of AKAP121 in mitochondrial physiology.Am J Physiol Cell Physiol. 2016 Apr 15;310(8):C625-8. doi: 10.1152/ajpcell.00292.2015. Epub 2016 Jan 28. Am J Physiol Cell Physiol. 2016. PMID: 26825124 Free PMC article. Review.
Cited by
-
A-Kinase Anchoring Protein 1: Emerging Roles in Regulating Mitochondrial Form and Function in Health and Disease.Cells. 2020 Jan 26;9(2):298. doi: 10.3390/cells9020298. Cells. 2020. PMID: 31991888 Free PMC article. Review.
-
Role of A-Kinase Anchoring Protein 1 in Retinal Ganglion Cells: Neurodegeneration and Neuroprotection.Cells. 2023 Jun 3;12(11):1539. doi: 10.3390/cells12111539. Cells. 2023. PMID: 37296658 Free PMC article. Review.
-
Coupling axonal mRNA transport and local translation to organelle maintenance and function.Curr Opin Cell Biol. 2022 Feb;74:97-103. doi: 10.1016/j.ceb.2022.01.008. Epub 2022 Feb 24. Curr Opin Cell Biol. 2022. PMID: 35220080 Free PMC article. Review.
-
Phosphoregulation on mitochondria: Integration of cell and organelle responses.CNS Neurosci Ther. 2019 Jul;25(7):837-858. doi: 10.1111/cns.13141. Epub 2019 Apr 25. CNS Neurosci Ther. 2019. PMID: 31025544 Free PMC article. Review.
-
Mitochondrial PKA Is Neuroprotective in a Cell Culture Model of Alzheimer's Disease.Mol Neurobiol. 2021 Jul;58(7):3071-3083. doi: 10.1007/s12035-021-02333-w. Epub 2021 Feb 23. Mol Neurobiol. 2021. PMID: 33624140 Free PMC article.
References
-
- T.Coyle J, and Puttfarcken P (1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science 262(5134), 689–695. - PubMed
-
- Gardoni F, and Di Luca M (2015) Targeting glutamatergic synapses in Parkinson’s disease. Current opinion in pharmacology 20, 24–28 - PubMed
-
- Murphy TH, Miyamoto M, Sastre A, Schnaar RL, and Coyle JT (1989) Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2, 1547–1558 - PubMed
-
- Fukui M, Song JH, Choi J, Choi HJ, and Zhu BT (2009) Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur J Pharmacol 617, 1–11 - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
