

Clinical and biological features in PIEZO1-hereditary xerocytosis and Gardos channelopathy: a retrospective series of 126 patients
- PMID: 30655378
- PMCID: PMC6669138
- DOI: 10.3324/haematol.2018.205328
Clinical and biological features in PIEZO1-hereditary xerocytosis and Gardos channelopathy: a retrospective series of 126 patients
Abstract
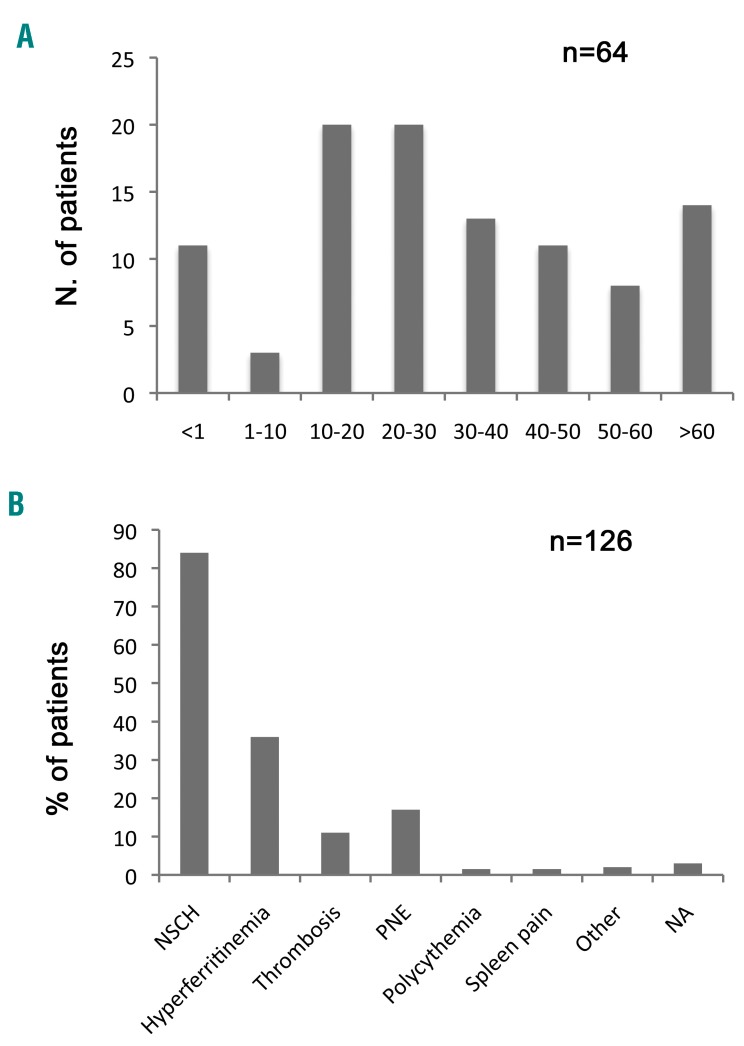
We describe the clinical, hematologic and genetic characteristics of a retrospective series of 126 subjects from 64 families with hereditary xerocytosis. Twelve patients from six families carried a KCNN4 mutation, five had the recurrent p.Arg352His mutation and one had a new deletion at the exon 7-intron 7 junction. Forty-nine families carried a PIEZO1 mutation, which was a known recurrent mutation in only one-third of the cases and private sequence variation in others; 12 new probably pathogenic missense mutations were identified. The two dominant features leading to diagnosis were hemolysis that persisted after splenectomy and hyperferritinemia, with an inconstant correlation with liver iron content assessed by magnetic resonance imaging. PIEZO1-hereditary xerocytosis was characterized by compensated hemolysis in most cases, perinatal edema of heterogeneous severity in more than 20% of families and a major risk of post-splenectomy thrombotic events, including a high frequency of portal thrombosis. In KCNN4-related disease, the main symptoms were more severe anemia, hemolysis and iron overload, with no clear sign of red cell dehydration; therefore, this disorder would be better described as a 'Gardos channelopathy'. These data on the largest series to date indicate that PIEZO1-hereditary xerocytosis and Gardos channelopathy are not the same disease although they share hemolysis, a high rate of iron overload and inefficient splenectomy. They demonstrate the high variability in clinical expression as well as genetic bases of PIEZO1-hereditary xerocytosis. These results will help to improve the diagnosis of hereditary xerocytosis and to provide recommendations on the clinical management in terms of splenectomy, iron overload and pregnancy follow-up.
Copyright© 2019 Ferrata Storti Foundation.
Figures


References
-
- Caulier A, Rapetti-Mauss R, Guizouarn H, Picard V, Garçon L, Badens C. Primary red cell hydration disorders: pathogenesis and diagnosis. Int J Lab Hematol. 2018;40 Suppl 1:68–73. - PubMed
-
- Delaunay J. The hereditary stomatocytoses: genetic disorders of the red cell membrane permeability to monovalent cations. Semin Hematol. 2004;41(2):165–172. - PubMed
-
- Andolfo I, Russo R, Gambale A, Iolascon A. Hereditary stomatocytosis: an underdiagnosed condition. Am J Hematol. 2018;93(1): 107–121. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
