

Autophagy-lysosome pathway alterations and alpha-synuclein up-regulation in the subtype of neuronal ceroid lipofuscinosis, CLN5 disease
- PMID: 30655561
- PMCID: PMC6336884
- DOI: 10.1038/s41598-018-36379-z
Autophagy-lysosome pathway alterations and alpha-synuclein up-regulation in the subtype of neuronal ceroid lipofuscinosis, CLN5 disease
Abstract
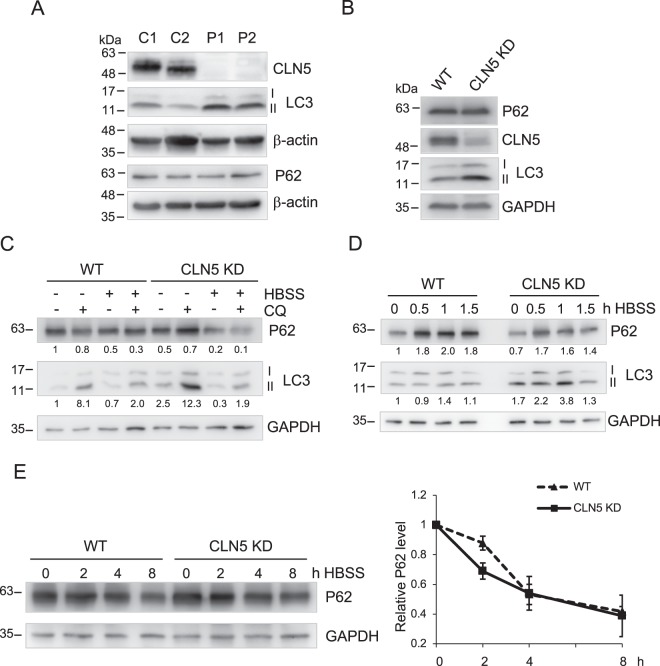
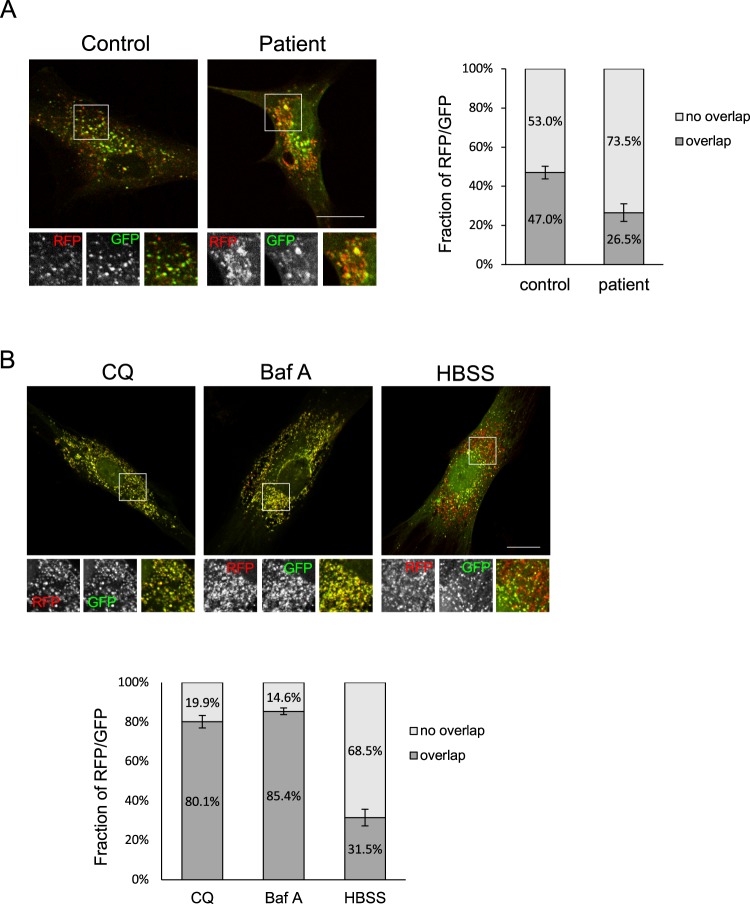
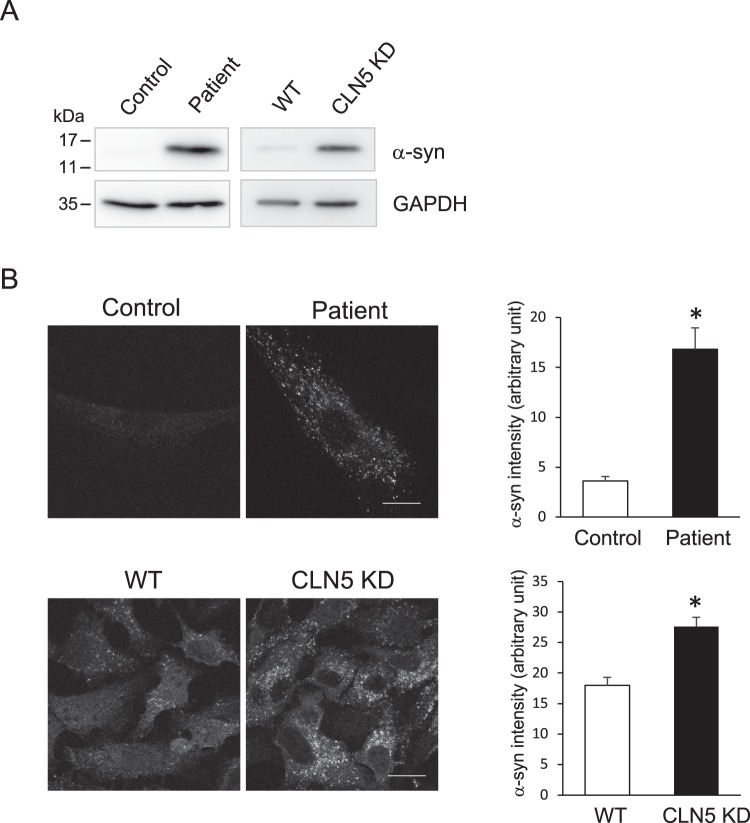
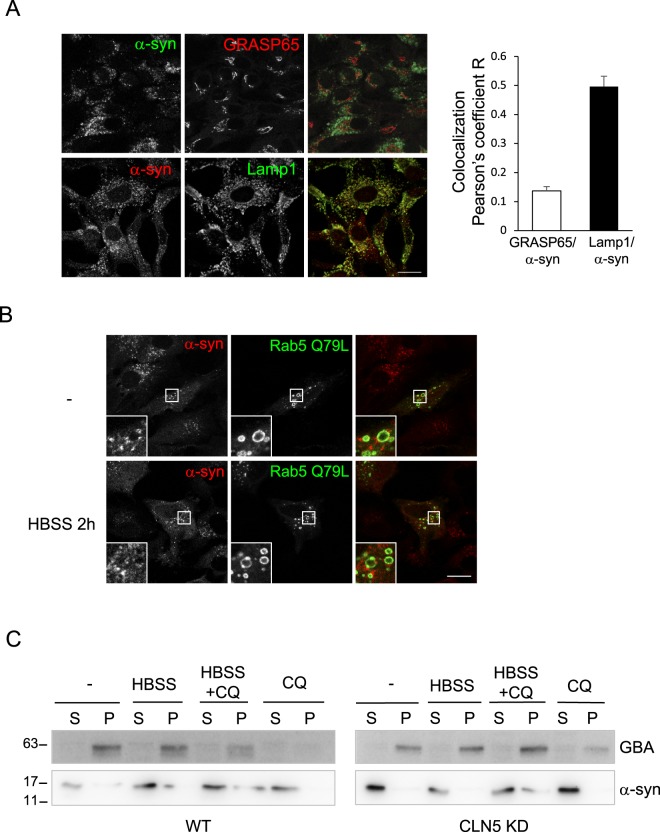
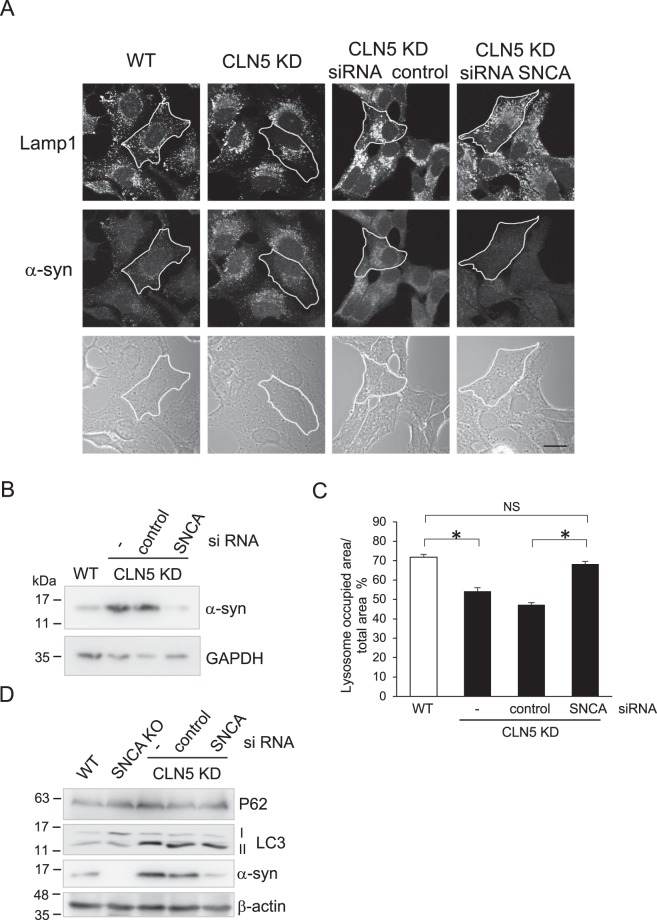
Neuronal ceroid lipofuscinoses (NCLs) are a group of inherited neurodegenerative lysosomal storage disorders. CLN5 deficiency causes a subtype of NCL, referred to as CLN5 disease. CLN5 is a soluble lysosomal protein with an unclear function in the cell. Increased levels of the autophagy marker protein LC3-II have been reported in several subtypes of NCLs. In this report, we examine whether autophagy is altered in CLN5 disease. We found that the basal level of LC3-II was elevated in both CLN5 disease patient fibroblasts and CLN5-deficient HeLa cells. Further analysis using tandem fluorescent mRFP-GFP-LC3 showed the autophagy flux was increased. We found the alpha-synuclein (α-syn) gene SNCA was highly up-regulated in CLN5 disease patient fibroblasts. The aggregated form of α-syn is well known for its role in the pathogenicity of Parkinson's disease. Higher α-syn protein levels confirmed the SNCA up-regulation in both patient cells and CLN5 knockdown HeLa cells. Furthermore, α-syn was localized to the vicinity of lysosomes in CLN5 deficient cells, indicating it may have a lysosome-related function. Intriguingly, knocking down SNCA reversed lysosomal perinuclear clustering caused by CLN5 deficiency. These results suggest α-syn may affect lysosomal clustering in non-neuronal cells, similar to its role in presynaptic vesicles in neurons.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
