

Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins
- PMID: 30655609
- PMCID: PMC7325303
- DOI: 10.1038/s41580-018-0089-8
Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins
Abstract
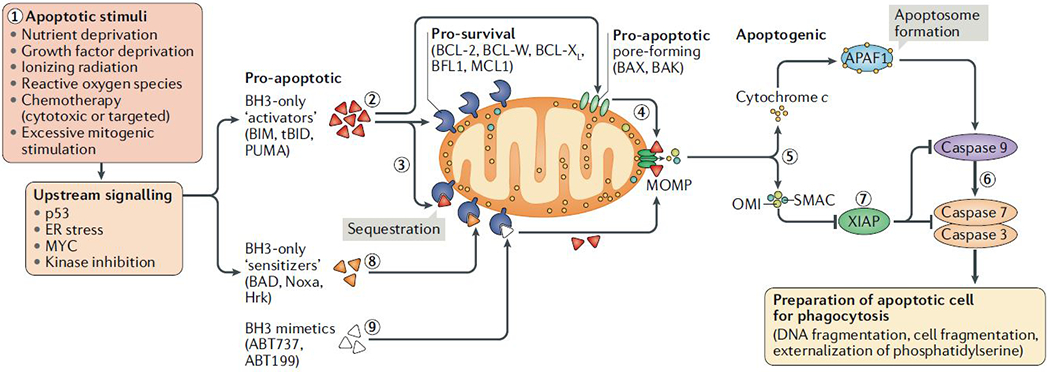
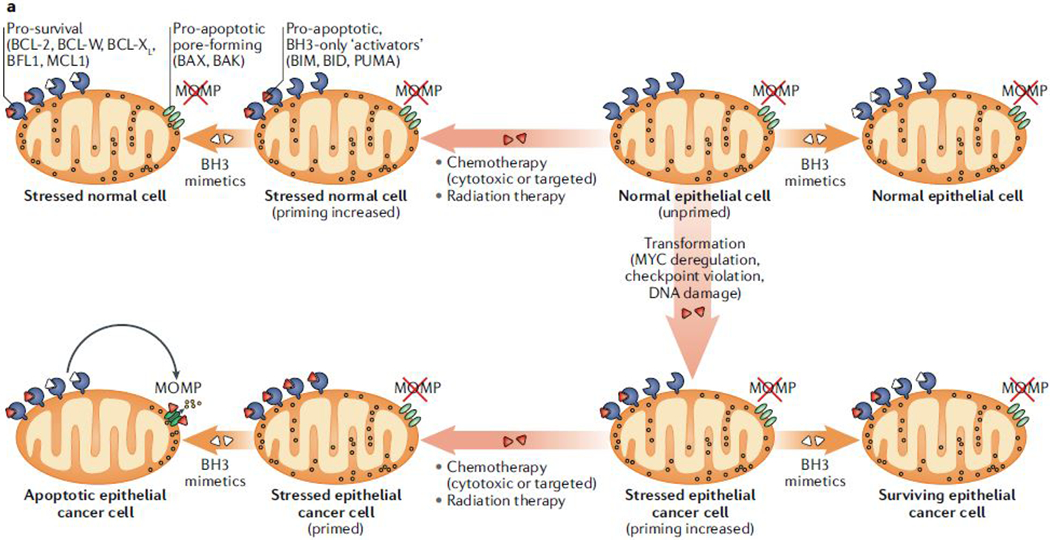
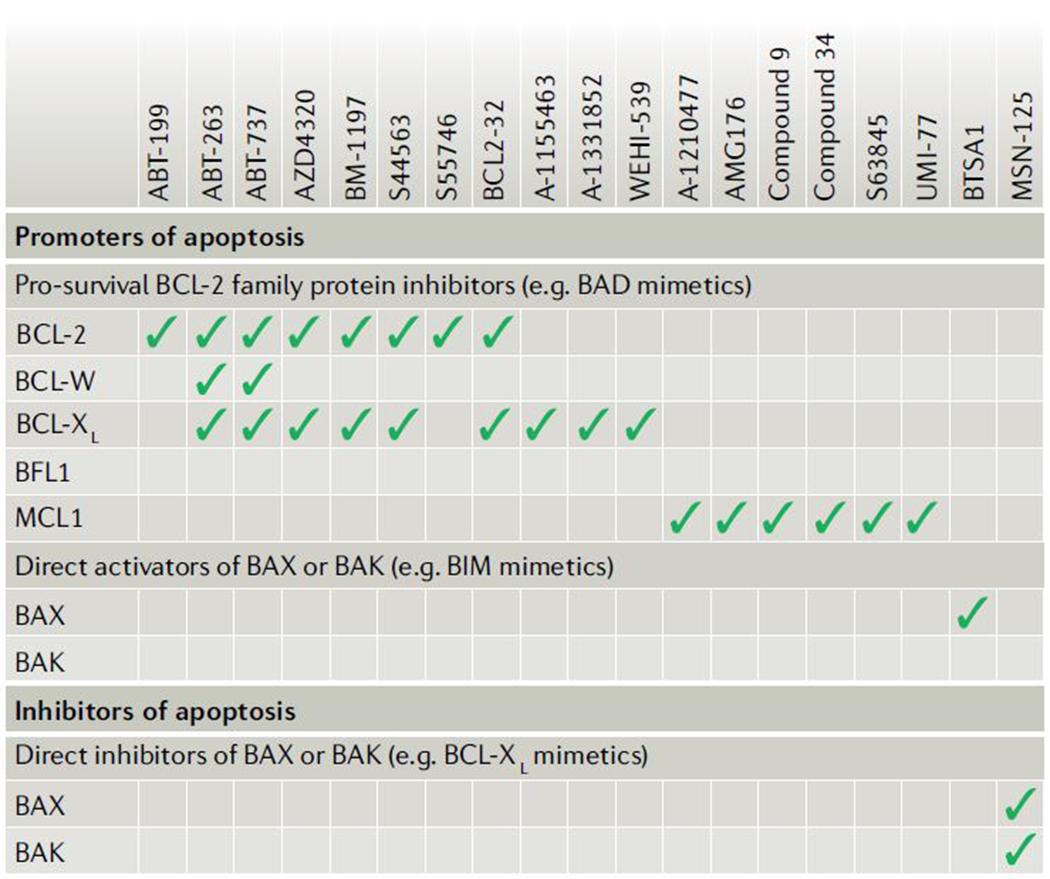
The loss of vital cells within healthy tissues contributes to the development, progression and treatment outcomes of many human disorders, including neurological and infectious diseases as well as environmental and medical toxicities. Conversely, the abnormal survival and accumulation of damaged or superfluous cells drive prominent human pathologies such as cancers and autoimmune diseases. Apoptosis is an evolutionarily conserved cell death pathway that is responsible for the programmed culling of cells during normal eukaryotic development and maintenance of organismal homeostasis. This pathway is controlled by the BCL-2 family of proteins, which contains both pro-apoptotic and pro-survival members that balance the decision between cellular life and death. Recent insights into the dynamic interactions between BCL-2 family proteins and how they control apoptotic cell death in healthy and diseased cells have uncovered novel opportunities for therapeutic intervention. Importantly, the development of both positive and negative small-molecule modulators of apoptosis is now enabling researchers to translate the discoveries that have been made in the laboratory into clinical practice to positively impact human health.
Figures




References
-
- Rathmell JC & Thompson CB Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell 109, S97–107 (2002). - PubMed
-
- Lamhamedi-Cherradi SE, Zheng SJ, Maguschak KA, Peschon J & Chen YH Defective thymocyte apoptosis and accelerated autoimmune diseases in TRAIL−/− mice. Nat. Immunol 4, 255–260 (2003). - PubMed
-
- Su JH, Deng G & Cotman CW Bax Protein Expression Is Increased in Alzheimer’s Brain: Correlations with DNA Damage, Bcl-2 Expression, and Brain Pathology. J. Neuropathol. Exp. Neurol 56, 86–93 (1997). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
