

Essential metabolism for a minimal cell
- PMID: 30657448
- PMCID: PMC6609329
- DOI: 10.7554/eLife.36842
Essential metabolism for a minimal cell
Abstract
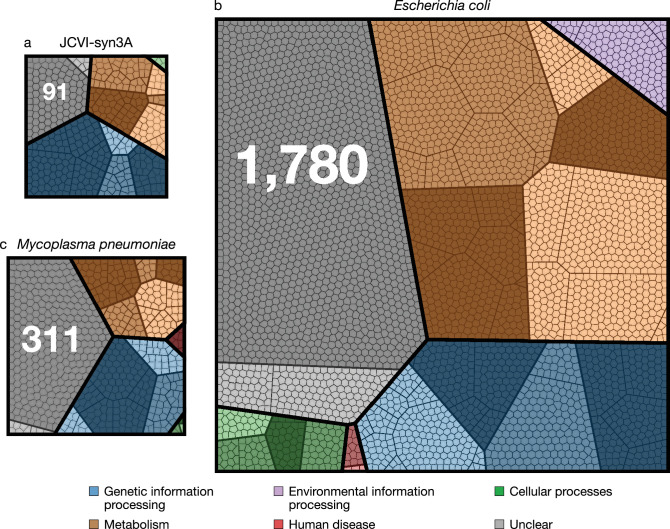
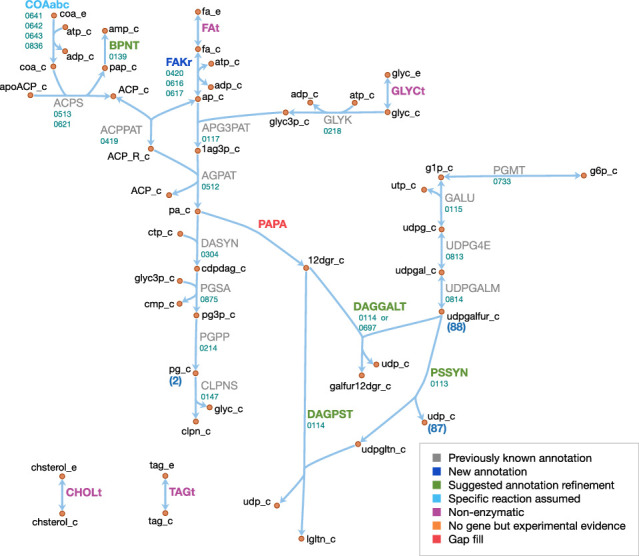
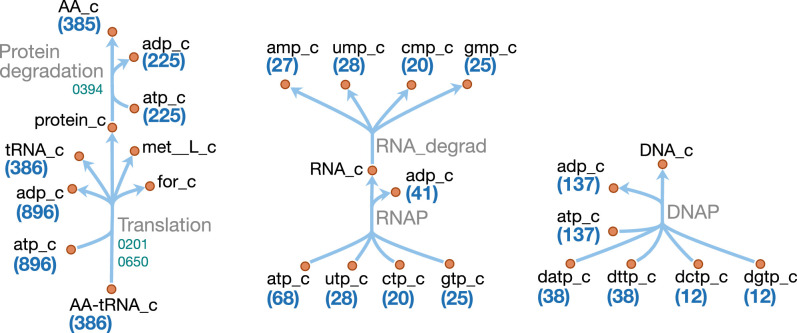
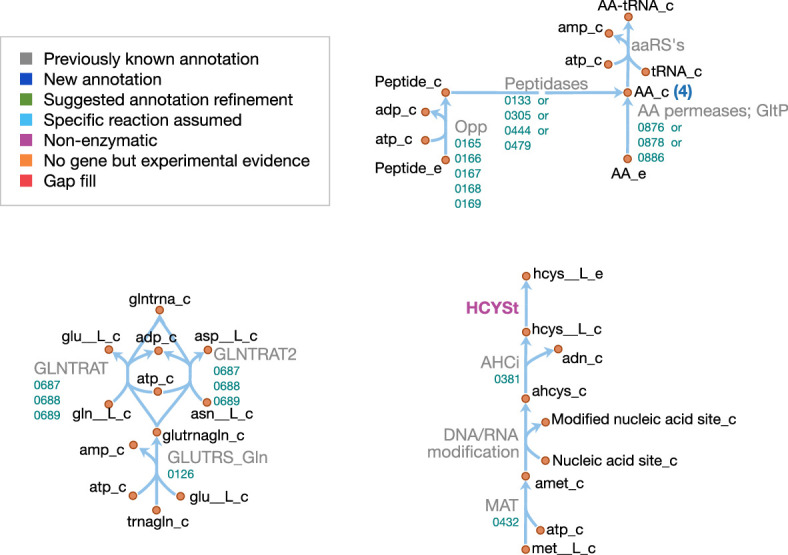
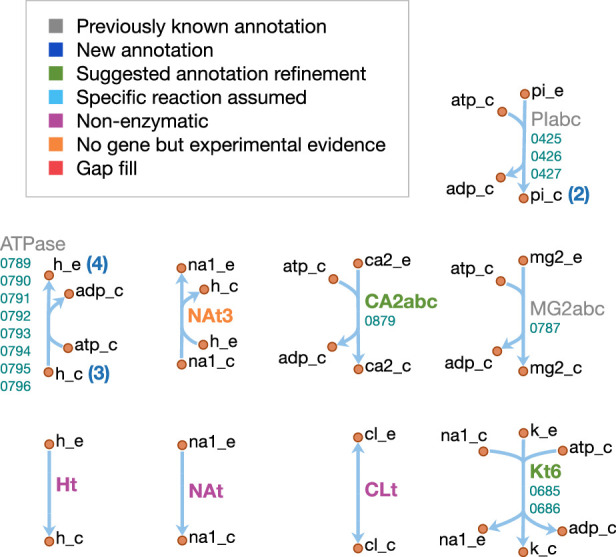
JCVI-syn3A, a robust minimal cell with a 543 kbp genome and 493 genes, provides a versatile platform to study the basics of life. Using the vast amount of experimental information available on its precursor, Mycoplasma mycoides capri, we assembled a near-complete metabolic network with 98% of enzymatic reactions supported by annotation or experiment. The model agrees well with genome-scale in vivo transposon mutagenesis experiments, showing a Matthews correlation coefficient of 0.59. The genes in the reconstruction have a high in vivo essentiality or quasi-essentiality of 92% (68% essential), compared to 79% in silico essentiality. This coherent model of the minimal metabolism in JCVI-syn3A at the same time also points toward specific open questions regarding the minimal genome of JCVI-syn3A, which still contains many genes of generic or completely unclear function. In particular, the model, its comparison to in vivo essentiality and proteomics data yield specific hypotheses on gene functions and metabolic capabilities; and provide suggestions for several further gene removals. In this way, the model and its accompanying data guide future investigations of the minimal cell. Finally, the identification of 30 essential genes with unclear function will motivate the search for new biological mechanisms beyond metabolism.
Keywords: JCVI-syn3A; computational biology; gene essentiality; metabolic reconstruction; mycoplasma; proteomics; systems biology; transposon mutagenesis.
Plain language summary
One way that researchers can test whether they understand a biological system is to see if they can accurately recreate it as a computer model. The more they learn about living things, the more the researchers can improve their models and the closer the models become to simulating the original. In this approach, it is best to start by trying to model a simple system. Biologists have previously succeeded in creating ‘minimal bacterial cells’. These synthetic cells contain fewer genes than almost all other living things and they are believed to be among the simplest possible forms of life that can grow on their own. The minimal cells can produce all the chemicals that they need to survive – in other words, they have a metabolism. Accurately recreating one of these cells in a computer is a key first step towards simulating a complete living system. Breuer et al. have developed a computer model to simulate the network of the biochemical reactions going on inside a minimal cell with just 493 genes. By altering the parameters of their model and comparing the results to experimental data, Breuer et al. explored the accuracy of their model. Overall, the model reproduces experimental results, but it is not yet perfect. The differences between the model and the experiments suggest new questions and tests that could advance our understanding of biology. In particular, Breuer et al. identified 30 genes that are essential for life in these cells but that currently have no known purpose. Continuing to develop and expand models like these to reproduce more complex living systems provides a tool to test current knowledge of biology. These models may become so advanced that they could predict how living things will respond to changing situations. This would allow scientists to test ideas sooner and make much faster progress in understanding life on Earth. Ultimately, these models could one day help to accelerate medical and industrial processes to save lives and enhance productivity.
© 2019, Breuer et al.
Conflict of interest statement
MB, EE, CM, KW, LS, ML, JL, DG, Vd, DH, AH, PL, JG, ZL No competing interests declared, CH is a consultant for Synthetic Genomics, Inc. (SGI), and holds SGI stock and/or stock options, HS is on the Board of Directors and cochief scientific officer of Synthetic Genomics, Inc. (SGI) and holds SGI stock and/or stock options
Figures
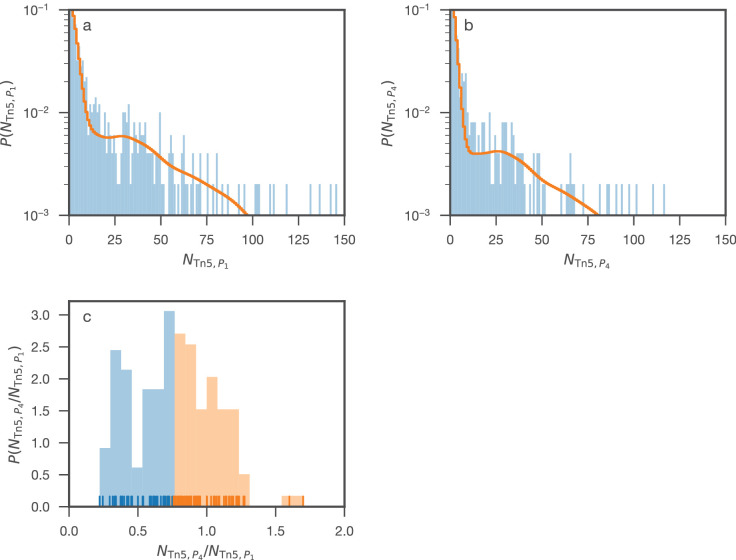
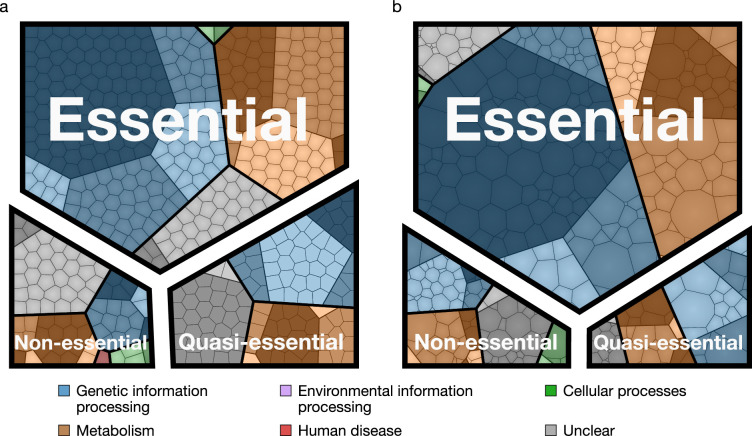

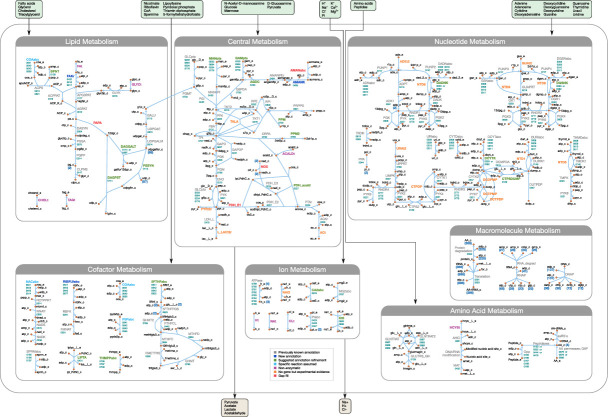
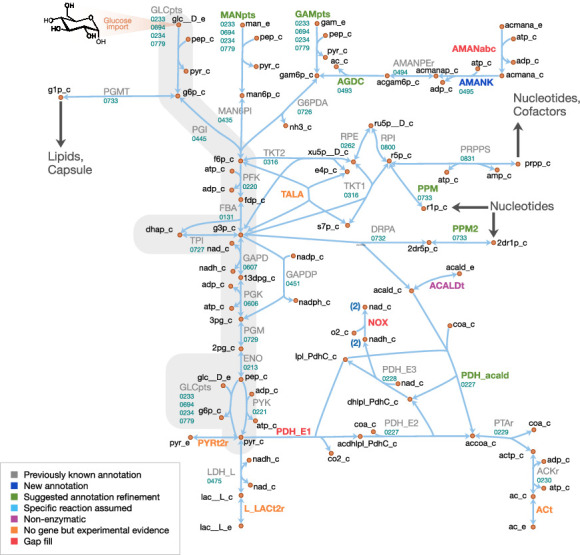
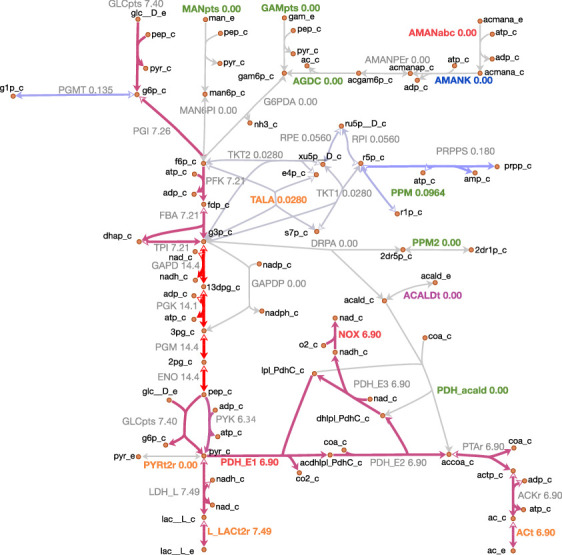
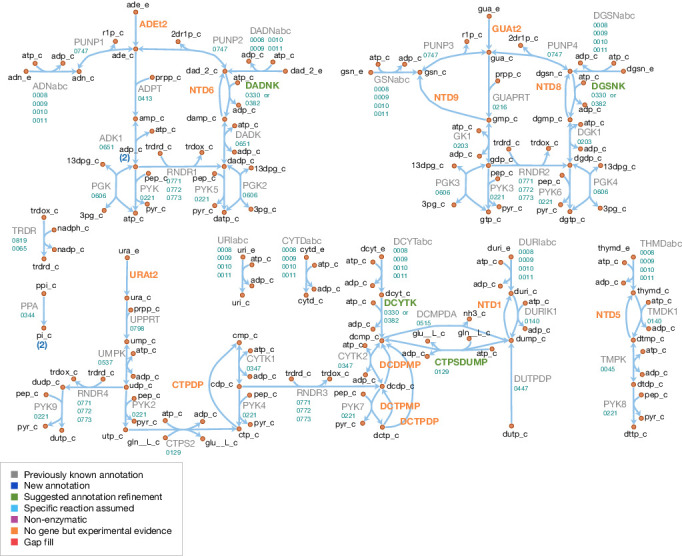
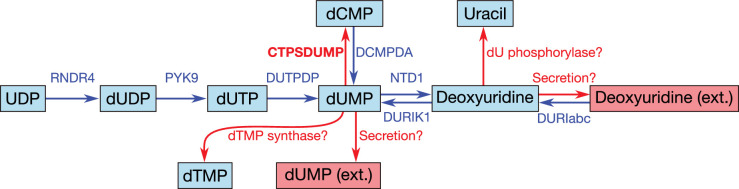
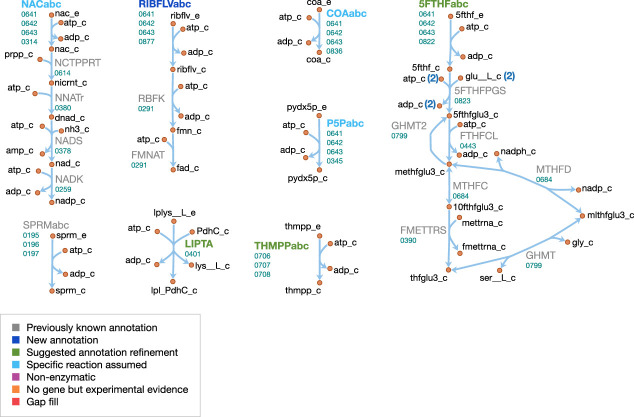
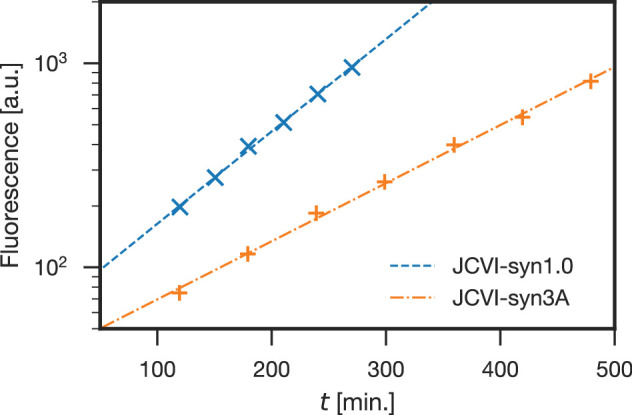
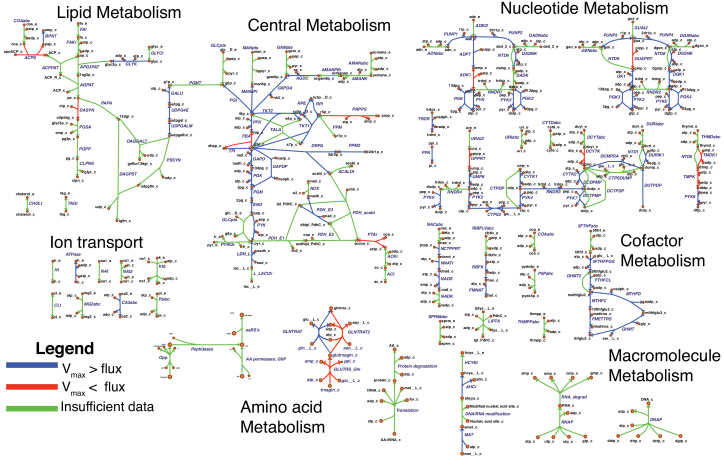
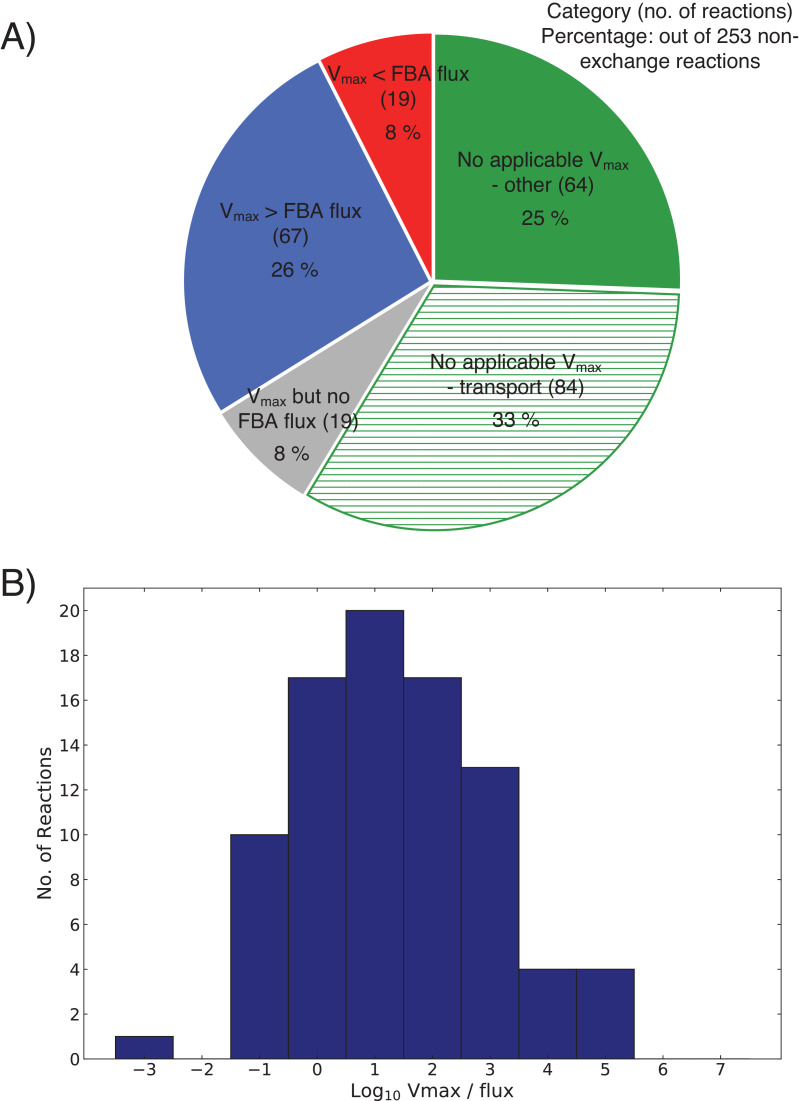
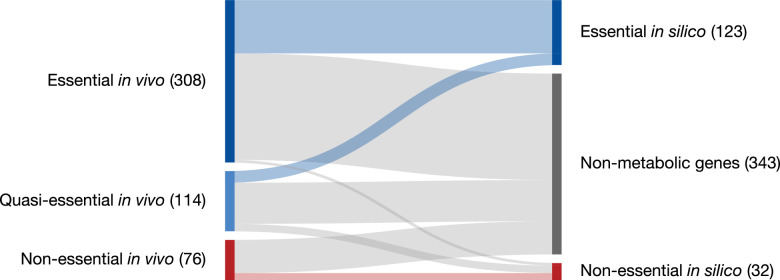

Comment in
- doi: 10.7554/eLife.45379
References
-
- Andrés E, Martínez N, Planas A. Expression and characterization of a Mycoplasma genitalium glycosyltransferase in membrane glycolipid biosynthesis: potential target against Mycoplasma infections. The Journal of Biological Chemistry. 2011;286:35367–35379. doi: 10.1074/jbc.M110.214148. - DOI - PMC - PubMed
-
- Archer D. Modification of the membrane composition of Mycoplasma mycoides subsp. capri by the growth medium. Microbiology. 1975;88:329–338. - PubMed
-
- Arora S, Bhamidimarri SP, Bhattacharyya M, Govindan A, Weber MH, Vishveshwara S, Varshney U. Distinctive contributions of the ribosomal P-site elements m2G966, m5C967 and the C-terminal tail of the S9 protein in the fidelity of initiation of translation in Escherichia coli. Nucleic Acids Research. 2013;41:4963–4975. doi: 10.1093/nar/gkt175. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
